| [Molecules: 12] [Related articles/posters: 112 083 015 054 114 ] |
For this reason, we decided to study their behaviour towards diazomethane, and we wish to report that these reactions are not only completely regioselective (as already known in the literature; refs 2,3, but are often highly diastereoselective as well.
Our results apparently were not in agreement with the well known inside alkoxy theory 4, since the syn diastereoisomer, and not the anti, is preferred. A rationale was found within the framework of the same effects on which the inside alkoxy theory is based.
References and experimental details are given at the end of the paper.
These cycloadducts were not subject to isomerization because they lack an acidic hydrogen; a different situation was found in the reactions of (Z)- and (E)-alkenes. The relative configuration of cycloadduct syn-5 was assigned by chemical correlation to the bicyclic lactone 7. NMR analysis revealed a NOE effect betweeh H-4 and H-4', that can be accounted for only by a cis configuration.

This is also in agreement with known literature data for diazomethane cycloadditions on similar compounds 5.
When the reaction mixture was irradiated (sunlight is enough), syn-5 decomposes to the corresponding cyclopropane derivative 8 as a single diastereoisomer, in 91% yield; its configuration was not assigned, and is indicated only tentatively as 2,2'-syn.
Syn-4 and syn-6 did not decompose under these conditions and were recovered in good chemical yields (>88%). To verify if the complete stereoselectivity in these reactions was due to the steric requirements (1-3 are (Z)-alkenes) and not to the presence of two electron withdrawing groups on the olefin, we performed diazomethane cycloadditions to the corresponding monoactivated (Z)-alkenes.
The diazomethane cycloaddition to (Z)-alkenes was again completely stereoselective; in analogy with our previous results on doubly activated alkenes and with literature data5, a syn configuration was assigned to all pyrazolines.
All cycloaddition products rapidly isomerized because of the presence of an acidic hydrogen a to the ester group: only d-2 pyrazolines were isolated in good yields from the reaction mixture.
In the presence of sunlight, the reaction on (Z)-9 gave pyrazoline syn-12 in quantitative chemical yield and complete stereoselectivity. The adducts derived from (Z)-10 and (Z)-11 decomposed under these conditions to unidentified products.
Thus the complete syn stereoselectivity must be due to the (Z) configuration at the double bond.
What happened with sterically unhindered (E)-alkenes?
With (E)-alkenes the stereochemical outcome of the reaction is more complicated. (E)-alkenes derived from lactaldehyde react with low 4,4'-syn selectivity.
The configuration of the major isomer was assigned by analogy with our previous data.
(E)-9, bearing an OBn residue in the allylic position, is slightly less selective (syn/anti = 67/33) than the corresponding OTBS derivative (E)-10 (syn/anti = 70/30).
d-1 pyrazolines 15 and 16 did not isomerize to the more stable d-2 pyrazolines. Since the alkene geometry is retained in these reactions, we assigned the anti relative configuration at the stereocentres at C-3 and C-4.
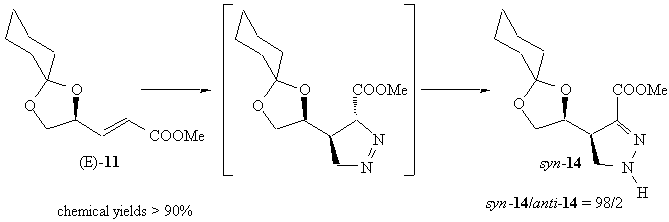
On the other hand, the reaction of (E)-11, derived from gliceraldehyde, was completely 4,4'-syn stereoselective; the cycloaddition product isomerized completely to the corresponding d-2 pyrazoline. Thus, from (E)-11 and (Z)-11 the same product (syn-14) was obtained.
With (E)-alkenes, the stereoselectivity depends on the substituent at the allylic oxygen and on the presence of a second oxygen in the homoallylic position. The same trend was observed for nitrile oxide cycloadditions on monosubstituted and (E) alkenes, and was well rationalized by the inside alkoxy theory.
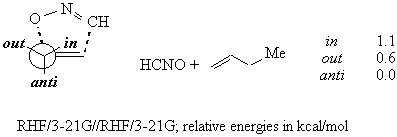
An alkoxyl residue avoids the anti position for stereoelectronic reasons.4
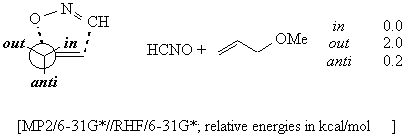
Within the inside and the outside positions, the latter is disfavoured because of the electrostatic repulsion between the two oxygens, one on the allylic stereocentre and one on the nitrile oxide. This repulsion is due to the amount of negative charge that is present on the nitrile oxide moiety in the transition structure. 6

How is the diazomethane charged in the transition structure?
Atomic charges in the TS for the diazomethane plus ethylene cycloaddition.

The terminal atoms on the diazomethane moiety in the transition structure for the cycloaddition reaction are not strongly charged.6 Thus there are no electrostatic interactions destabilizing the outside region for an alkoxy group on the allylic stereocentre.
The correlation between the amount of negative charge on the 1,3-dipole and the reaction stereoselectivity was demonstrated by previous studies.1
On this basis, we feel that a rationale can be proposed to account for the stereochemical outcome of the cycloaddition reactions between diazomethane and g-alkoxy-a, b-unsaturated esters
(Z)-alkenes:
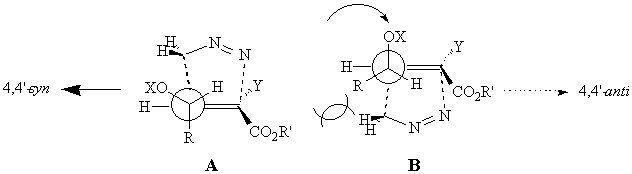
For these reactions, the inside position is the most congested and can be occupied only by the allylic hydrogen. Only two conformations are allowed for the TS, leading to formation of the 4,4'-syn (TS A) and 4,4'-anti (TS B) adducts; the former (TS A) features all the substituents at the allylic stereocentre in their preferred position, and is strongly favoured over the latter, that brings the same substituents into their disfavoured positions.
(E)-alkenes:
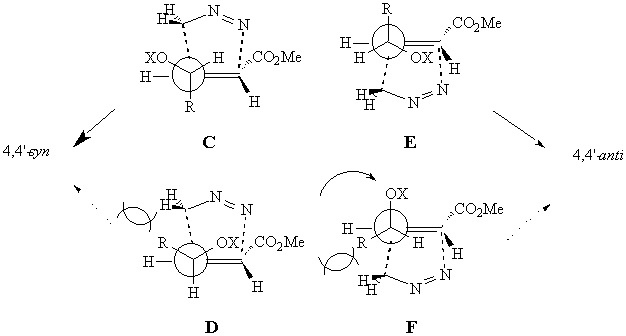
The conformational freedom at the allylic stereocentre allows also the alkoxy group to occupy the inside position. Four transition structures for the formation of the 4,4'-syn (TS C and D) and 4,4'-anti (TS E and F) adducts, can be considered. The TS conformations that bring the alkyl residue in the strongly disfavoured inside region will not be taken into account.
For the formation of the the 4,4'-syn isomers, TS C is probably the most stable, since the alkyl residue occupies the anti position while the alkoxy group avoids it.
For the formation of the the 4,4'-anti isomers, TS E is operating for the same reasons.
The low syn selectivity observed in the reaction of (E)-alkenes is probably due to a destabilization of TS E with respect to C because of the allylic strain that destabilizes the inside position.7
All reactions were run under nitrogen. Et2O was distilled from LiAlH4 and stored on molecular sieves. Purification of compounds by flash chromatography was performed with 230-400 mesh silica gel. Diazomethane was prepared as ethereal solution from DIAZALD and KOH in ethanol9 in the required amount, and the excess was immediately destroyed with AcOH. All reactions employing diazomethane were performed under a safety hood. IR spectra were recorded as liquid film. 1-H and 13-C NMR spectra were recorded on a Bruker AM 300 spectrometer at 300.133 and 75.47 MHz respectively, in CDCl3 as solvent; the 13-C {1-H} spectra were obtained using WALTZ decoupling. Chemical shifts are reported in d relative to SiMe4. For the 2D NOESY NMR experiment the solution was degassed by several freeze-pump-thaw cycles on a high-vacuum line to remove dissolved oxygen. Alkenes 1 and 210 and (E)- and (Z)-9-1111, 12 are known compounds. 3 was synthesized accordingly to the procedure reported for 2 10 and purified by flash chromatography (hexanes/Et2O = 85/15; 54%). IR: 3020, 2900, 1700 cm-1. Anal Calcd for C216H24O6: C, 61.52; H, 7.74. Found: C, 61.58; H, 7.70. 1H NMR (80 MHz) d: 1.28 (3H, t,J= 6.9 Hz), 1.32 (3H, t,J= 6.9 Hz), 1.50 (10H, m), 3.69 (1H, m), 4.30 (4H, m), 4.89 (2H, q,J= 6.9 Hz), 6.95 (1H, d,J= 7.0 Hz). [a]D = +10.23 (c 1.12, 20 °C, CHCl3).
General procedure for the diazomethane cycloaddition. To a stirred solution of diazomethane9 (5 mmol) in Et2O (15 ml), kept under nitrogen at 0 °C, the desired alkene was added (0.5 mmol), and the solution was kept at that temperature until completion of the reaction (1-4 h; monitored by TLC). When desired, the reaction was performed in the dark. In the synthesis of adducts syn-4-6 and of cyclopropane 8 only, the excess of diazomethane was destroyed by addition of AcOH (5 mmol). The solvent was removed under reduced pressure. Chemical yields and diastereoisomeric ratios are collected in Tables 1 and 2; 1-H and 13-C selected NMR data for cycloadducts syn-4-6 and 12-16 are collected in Table 3. Pyrazolines 12-16 were unstable at light, so no [a]D were measured.
3,3-Dicarbethoxy-4-[1'-(phenylmethoxy)ethyl]-1-pyrazoline syn-4: IR: 3000, 2850, 1730, 1550 cm-1. Anal Calcd for C18H24O5N2: C, 62.05; H, 6.94; N, 8.04. Found: C, 62.15; H, 6.92; N, 8.01. 1-H NMR: 2.83 (H-4); 3.88 (H-4'); 4.70, 4.33 (H-5). 13-C NMR: 100.8 (C-3); 44.5 (C-4); 71.0 (C-4'); 69.7 (C-5). [a]D = +144.4 (c 0.43, 20 °C, CHCl3).
3,3-Dicarbethoxy-4-[1'-[(1,1-dimethylethyl)dimethylsilyloxy]ethyl]-1-pyrazoline syn-5: IR: 2900, 1750, 1550 cm-1. Anal Calcd for C17H32O5N2Si: C, 54.81; H, 8.66; N, 7.52. Found: C, 54.78; H, 8.64; N, 7.55. 1-H NMR: 2.73 (H-4); 3.88 (H-4'); 4.65 (H-5). 13-C NMR: 106.0 (C-3); 46.0 (C-4); 80.4 (C-4'); 67.3 (C-5). [a]D = +151.2 (c 0.42, 20 °C, CHCl3).
3,3-Dicarbethoxy-4-[(1,4-dioxaspiro)dec-2-yl]-1-pyrazoline syn-6: IR: 2750, 1735, 1580 cm-1. Anal Calcd for C17H26O6N2: C, 57.61; H, 7.39; N, 7.90. Found: C, 57.65; H, 7.42; N, 7.85. 1-H NMR: 2.84 (H-4); 4.07(H-4'); 4.80, 4.57 (H-5). 13-C NMR: 106.0 (C-3); 41.2 (C-4); 73.0 (C-4'); 68.0 (C-5). [a]D = -139.7 (c 3.23, 20 °C, CHCl3).
3-Carbomethoxy-4-[1'-(phenylmethoxy)ethyl]-2-pyrazoline syn-12: IR: 3400, 2900, 1740, 1650 cm-1. Anal Calcd for C14H18O3N2: C, 64.10; H, 6.92; N, 10.68. Found: C, 64.07; H, 6.94; N, 10.65. 1-H NMR: 3.33 (H-4); 4.17(H-4'); 3.82, 3.64 (H-5). 13-C NMR: 142.45 (C-3); 50.3 (C-4); 71.9 (C-4'); 49.2 (C-5).
3-Carbomethoxy-4-[1'-[((1,1-dimethylethyl)dimethylsilyl)oxy]ethyl]-2-pyrazoline syn-13: IR: 3450, 2850, 1750, 1550 cm-1. Anal Calcd for C13H26O3N2Si: C, 54.51; H, 9.15; N, 9.78. Found: C, 54.45; H, 9.19; N, 9.85. 1-H NMR: 3.28 (H-4); 4.53 (H-4'); 3.85, 3.64 (H-5). 13-C NMR: 142.5 (C-3); 51.9 (C-4); 65.5 (C-4'); 48.0 (C-5).
3-Carbomethoxy-4-[(1,4-dioxaspiro)dec-2-yl]-2-pyrazoline syn-14: IR: 3400, 2900, 1730, 1600 cm-1. Anal Calcd for C13H20O4N2: C, 58.19; H, 7.51; N, 10.44. Found: C, 58.24; H, 7.47; N, 10.50. 1-H NMR: 3.38 (H-4); 4.36 (H-4'); 3.86, 3.67 (H-5). 13-C NMR: 141.0 (C-3); 51.4 (C-4); 73.6 (C-4'); 47.1 (C-5).
3-Carbomethoxy-4-[1'-(phenylmethoxy)ethyl]-1-pyrazolines syn- and anti-15: IR: 2950, 1740, 1555 cm-1. Anal Calcd for C14H18O3N2: C, 64.10; H, 6.92; N, 10.68. Found: C, 64.15; H, 6.90; N, 10.65. syn-15: 1-H NMR: 5.27 (H-3); 2.50 (H-4); 3.38 (H-4'); 4.67, 4.37 (H-5). 13-C NMR: 90.9 (C-3); 41.2 (C-4); 73.7 (C-4'); 70.1 (C-5). anti-15: 1-H NMR: 5.08 (H-3); 2.47 (H-4); 3.50 (H-4'); 4.58, 4.33 (H-5). 13-C NMR: 91.4 (C-3); 41.1 (C-4); 73.1 (C-4'); 70.6 (C-5).
3-Carbomethoxy-4-[1'-[((1,1-dimethylethyl)dimethylsilyl)oxy]ethyl]-1-pyrazolines syn- and anti-16: IR: 3400, 2850, 1750, 1600 cm-1. Anal Calcd for C13H26O3N2Si: C, 54.51; H, 9.15; N, 9.78. Found: C, 54.48; H, 9.21; N, 9.73. syn-16: 1-H NMR: 5.23 (H-3); 2.35 (H-4); 3.73 (H-4'); 4.60, 4.33 (H-5). 13-C NMR: 90.25 (C-3); 42.4 (C-4); 80.7 (C-4'); 67.9 (C-5). anti-16: 1-H NMR: 4.98 (H-3); 2.32 (H-4); 3.83 (H-4'); 4.52 (H-5). 13-C NMR: 91.65 (C-3); 42.8 (C-4); 77.4 (C-4'); 67.2 (C-5).
1,1-Dicarbethoxy-2-[1'-[((1,1-dimethylethyl)dimethylsilyl)oxy]ethyl]-cyclopropane 8 was purified by flash chromatography (hexanes/Et2O = 7/3). IR: 3050, 2950, 1750 cm-1. Anal Calcd for C17H32O5Si: C, 59.27; H, 9.36. Found: C, 59.30; H, 9.34. 1-H NMR (selected) d: 1.38 (H-3B, part B of an AB system,J= 9.9 Hz), 1.53 (H-3A, part A of an AB system,J= 9.9 Hz), 3.00 (H-2,J= 9.9; 9.0 Hz), 3.33 (H-2',J= 9.0; 6.5 Hz).13-C NMR (selected) d: 20.0 (C-3), 24.0 (CH3-2'), 33.9 (C-1), 35.2 (C-2), 68.4 (C-2'). [a]D = +29.9 (c 0.91, 20 °C, CHCl3).
Synthesis of 3-carbethoxy-8-methyl-2H,3H,7H,8H-tetrahydrofuro[3,4-c]-3H,4H-dihydropyrazole 7. To a stirred solution of syn-5 (0.2 mmol) in THF (5 ml), 18 ml of 40% HF were added, and the reaction was stirred at room temp. for 6 h. Solid NaHCO3 was added, and after 15 min of stirring the mixture was filtered; removal of solvent under reduced pressure gave 7 in 42% yield (determined by NMR). After flash chromatography (Et2O) a 5 mg sample pure enough for NOE experiments was recovered. The 1D and 2D NMR methods applied to perform the complete structural and stereochemical assignement of lactone 7, including inverse detection techniques, were the 13-C attached proton test, the heteronuclear multiple bond connectivity (HMBC) and the 2D NOESY sequences. For the HMBC experiment the following parameters were used: spectral width in f1 and f2 16000 and 1900 Hz, respectively, a 1024 x 1024 data matrix and 128 time increments of 512 transients each, relaxation delay of 2 s and optimum J value of 7.0 Hz. Pure absorption 2D NOESY experiment were recorded using the pulse sequence 90°-t1-90°-tm-90°-t2 and the TPPI method, described by Marion and Wüthrich,13 for the phase cycling. The parameters and procedure used were: spectral width of 2450 Hz, a 1024 x 1024 data matrix and 256 time increments of 48 transients each; mixing time of 1.5 s and relaxation delay of 6 s; Fourier trasformation was carried out with a shifted sine-bell apodization function in both dimensions. 1-H NMR (selected) d: 1.46 (CH3, J=6.5 Hz), 4.09 (H-4, J=6.5, 1.3 Hz), 4.95 (H-4', J=6.5, 6.0 Hz), 6.73 (H-5, J=1.3 Hz), 6.72 (NH). 13-C NMR (selected) d: 17.5 (CH3C-4'), 57.3 (C-4), 74.7 (C-3), 77.7 (C-4'), 139.0 (C-5), 173.0 (C-3').
1 Annunziata, R.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Raimondi, L. J. Org. Chem. 1995, 60, 4697
2 Regitz, M.; Heydt, H., in 1.3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley Interscience; New York, NY, 1984
3 Baskaran, S.; Vasu, J.; Prasad, R.; Kodkulla, K.; Trivedi, G. K.; Chandrasekhar, J. Tetrahedron 1996, 52, 4515
4 Houk, K. N.; Moses, S. R.; Wu, Y.-D.; Rondan, N. G.; Jäger, V.; Schohe, R.; Fronczek, F. R. J. Am. Chem. Soc. 1984, 106, 3880. Houk, K. N.; Dhu, H.-Y.; Wu, Y.-D.; Moses, S. R. J. Am. Chem. Soc. 1986, 108, 2754. Raimondi, L.; Wu, Y.-D-; Brown, F. K.; Houk, K. N. Tetrahedron Lett. 1992, 33, 4409
5 Galley, G.; Pätzel, M.; Jones, P. G. Tetrahedron 1995, 51, 1631
6 Annunziata, R.; Benaglia, M.; Cinquini, M.; Raimondi, L. Tetrahedron 1993, 49, 8629
7 Hoffmann, R. W. Chem. Rev. 1989 89, 841
8 For a recent on the diastereoselectivity in 1,3-dipolar cycloadditions, see: Cinquini, M.; Cozzi, F. Houben Weyl E21c; G. Thieme; Stuttgart, 1995, pg 2953.
9 Fieser, L. F.; Fieser, M. Reagents for Organic Synthesis. Wiley and Sons, Incorporated.
10 Yamamoto, Y.; Chounan, Y.; Nishii, S.; Ibuka, T.; Kitaharo, H. J. Am. Chem. Soc. 1992, 114, 7652.
11 Annunziata, R.; Cinquini, M.; Cozzi, F.; Dondio, G.; Raimondi, L. Tetrahedron 1987, 43, 2369.
12 Annunziata, R.; Cinquini, M.; Cozzi, F.; Raimondi, L.; Pilati, T. Tetrahedron: Asymmetry 1991, 2, 1329.
13 Marion, D.; Wüthrich, K. Biochem. Biophys. Res. Commun. 1983, 113, 967.