Vinylaziridines from vinyl epoxides
Iain Coldham,*a Alan J. Collisb and Roger J. Moulda
aDepartment of Chemistry, University of Exeter, Stocker Road, Exeter, UK EX4 4QD
bDepartment of Discovery Chemistry, Pfizer Central Research,
Sandwich, Kent, UK CT13 9NJ
Background
This paper reports a new method for the synthesis of aziridines containing an
alkene group attached to the 2-position of the aziridine ring.
2-Vinylaziridines1 are crucial intermediates for a number of
synthetic transformations. They have been used for the preparation of
pyrrolizidine2 and amaryllidaceae3 alkaloids, isosteres
as peptide mimetics4 and other biologically active
compounds.5
We were interested in preparing N-unsubstituted-2-vinylaziridines, as
substrates for our study of the aza-Wittig rearrangement (with concomitant ring
expansion) to tetrahydropyridines.6 Deprotonation a to the
nitrogen atom, exo to the aziridine ring, is followed by
[2,3]-sigmatropic rearrangement to the tetrahydropyridine ring.
We know very few methods for the preparation of
N-unsubstituted-2-vinylaziridines and would welcome feedback to Iain Coldham in this
area.
One method that we explored for the preparation of the required vinylaziridines
involved the simple two-step ring-opening of epoxides with azide, followed by
ring-closure to the aziridine with triphenylphosphine.7 The
extension of this reaction to the ring opening of vinylepoxides is reported
here.
Synthesis of vinyl epoxides
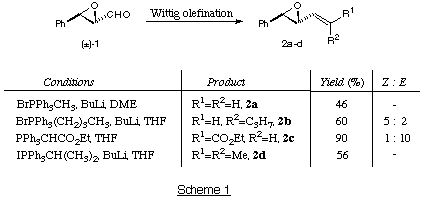
A selection of vinyl epoxides were prepared from cinnamaldehyde, according to
Scheme 1. Epoxidation of cinnamaldehyde was performed using hydrogen peroxide
in aqueous sodium bicarbonate.8 The epoxy-aldehyde was converted,
using Wittig olefination, to the required vinylepoxides. Four vinyl epoxides,
2a.9 2b, 2c10 and
2d11 with different substitution patterns around the alkene
were prepared. The epoxide 2b was formed as a mixture of stereoisomers,
favouring the Z-alkene in a ratio 5:2 (experimental and data). The
epoxide 2c was isolated as, predominantly, the more stable E
isomer (10:1).
Ring-opening of the vinyl epoxides
All four vinyl epoxides undergo smooth ring-opening with azide under standard
conditions7 (NaN3, NH4Cl, MeOH, H2O, heat) as shown in Scheme 2.
The expected b-azido-alcohols 3a-d and 4a-d are the major
products, with no significant regioselectivity in the ring-opening step. Both
the phenyl ring and the alkene group appear to have a similar influence on the
electrophilicity of the epoxide. The mixture of regioisomeric
b-azido-alcohols is not a problem, as both regioisomers can be taken on to
the next stage.
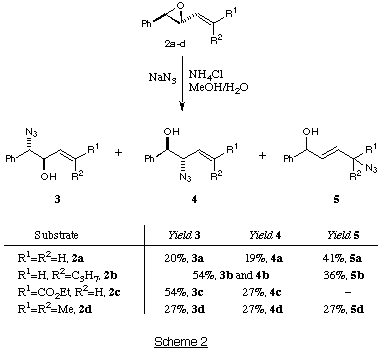
In addition to the b-azido-alcohols 3a-d and 4a-d, the
azido-alcohols 5 were isolated in all cases, except from vinyl epoxide
2c. The formation of 5a-b and 5d is interesting and could involve one (or both!) of two mechanisms.
Experimental conditions and data for azido-alcohols 3-5
Direct SN2' attack of the azide ion on to the vinyl epoxide would lead to the
products 5. Alternatively, [3,3]-sigmatropic shift of 4
via an allylic azide rearrangement would give 5. The likely
pathway followed is via the [3,3] pathway, as it is possible to isolate
and separate all three azido-alcohols 3a, 4a and 5a and
observe that the b-azido-alcohol 4a converts slowly at room
temperature to the d-azido-alcohol 5a. The extent of this
rearrangement is greatest when R=R'=H as this converts the monosubstituted
alkene into a disubstituted alkene. No rearrangement to the
d-azido-alcohol 5c was observed as this would take the double bond
out of conjugation with the ester functionality.10
Ring-closure with triphenylphosphine - Synthesis of vinylaziridines
The mixture of regioisomeric b-azido-alcohols 3a-d and 4a-d
can be ring-closed to the aziridines 6a-d using triphenylphosphine in
acetonitrile (Scheme 3).
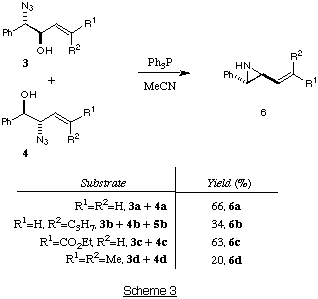
The b-azido-alcohols 3a and 4a were taken on immediately in
order to avoid the [3,3]-allylic azide shift. This gave the vinylaziridine
6a in reasonable yield (experimental).
For the formation of the aziridine 6b, the mixture of azido-alcohols
3b, 4b and 5b were used, although treatment of 5b
with Ph3P did not result in the formation of aziridine 6b. The ratio of
alkene stereoisomers of vinylaziridine 6b had changed to 3:2 in favour
of the E alkene (experimental). We suspect that this may be a result of
the preferential allylic azide rearrangement of Z-4b.
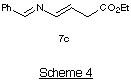
The ring closure with b-azido-alcohol 3c and 4c had to be
carried out at room temperature, otherwise the aziridine 6c was isolated
in low yield (36%), together with the imine 7c (35%) (Scheme
4).10 We have found that heating aziridine 6c in MeCN
results in the formation of 7c via an azo-methine ylide. This
ylide can be trapped with dipolarophiles to give pyrrolidine
product.10 The type of ring-opening that occurs with the aziridine
6c does not take place with the other vinylaziridines as it requires the
electron-withdrawing ester group to stabilise the ylide.
The aziridine 6d was isolated in low yield and was unstable to
purification. This is a result of the two methyl groups (R [1] and
R [2]), which presumably activate the aziridine to electrophilic
ring-opening (with build-up of positive charge stabilised by the two Me
groups).
Conclusions
The well-known methodology for the conversion of epoxides to aziridines
via azide opening, followed by closure with Ph3P has been shown to be
successful on a selection of vinyl epoxides. This has allowed a short
(three-steps from an epoxy-aldehyde) method for the preparation of
vinylaziridines. The N-unsubstituted-2-vinylaziridine 6c is
formed in 46% overall yield for the three steps from aldehyde 1. The
overall yield is reduced for other vinylaziridines, due mostly to problems with
allylic rearrangement of the azide group.
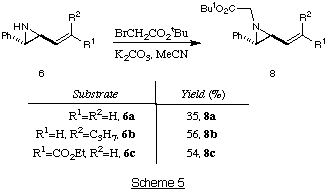
We have shown that such vinylaziridines can be functionalised further, by
alkylation on the nitrogen atom of the vinylaziridine, as shown in Scheme 5.
This sets the stage for the utilisation of these vinyl aziridines for further
transformations in organic synthesis.
Acknowledgements
We thank the SERC for an Earmarked Studentship (R.J.M.) and Pfizer Central
Research for a CASE Award (R.J.M.).
References
- D. Borel, Y. Gelas-Mialhe and R. Vessiere, Can. J. Chem., 1976,
54, 1590; J. Sauleau, A. Sauleau and J. Huet, Bull. Soc. Chim.
Fr., 1978, 97; A. Hassner and W. Chau, Tetrahedron Lett., 1982,
23, 1989; M.E.M. Attia, Y.Gelas-Mialhe and R. Vessiere, Can. J.
Chem., 1983, 61, 2126; W.H. Pearson, Tetrahedron Lett., 1985,
26, 3527; T. Hudlicky, J.O. Frazier, G. Seoane, M. Tiedje, A. Seoane,
L.D. Kwart and C. Beal, J. Am. Chem. Soc., 1986, 108, 3755; J.
Åhman, P. Somfai, and D. Tanner, J. Chem. Soc., Chem. Commun.,
1994, 2785; P. Somfai and J. Åhman, Tetrahedron Lett., 1995,
36, 1953; A. Satake, I. Shimizu and A. Yamamoto, Synlett., 1995,
64; J.G. Knight and M.P. Muldowney, Synlett., 1995, 949; N. Mimura, T.
Ibuka, M. Akaji, Y. Miwa, T. Taga, K. Nakai, H. Tamamura, N. Fujii and Y.
Yamamoto, Chem. Commun., 1996, 351; A.-H. Li, L.-X. Dai and X.-L Hou,
Chem. Commun., 1996, 491.
-
T. Hudlicky and J.W. Reed in 'Comprehensive Organic Synthesis', Ed. B.M.
Trost and I. Fleming, Pergamon, 1991, vol. 5, ch. 8.1, p. 899.
-
X. Tian, R. Maurya, K. Königsberger and T. Hudlicky, Synlett.,
1995, 1125; X. Tian, T. Hudlicky and K. Königsberger, J. Am. Chem.
Soc., 1995, 117, 3643.
-
P. Wipf and P.C. Fritch, J. Org. Chem., 1994, 59, 4875; N.
Fujii, K. Nakai, H. Tamamura, A. Otaka, N. Mimura, Y. Miwa, T. Taga, Y.
Yamamoto and T. Ibuka, J. Chem. Soc., Perkin Trans. 1, 1995, 1359.
-
G.W. Spears, K. Nakanishi and Y. Ohfune, Synlett., 1991, 91; D.
Tanner and P. Somfai, Bioorg. Med. Chem. Lett., 1993, 3, 2415.
-
I. Coldham, A.J. Collis, R.J. Mould and R.E. Rathmell, Tetrahedron
Lett., 1995, 36, 3557; J. Chem. Soc., Perkin Trans. 1, 1995,
2739. See also, J. Åhman and P. Somfai, J. Am. Chem. Soc., 1994,
116, 9781;Tetrahedron, 1995, 51, 9747.
-
Y. Ittah, Y. Sasson, I. Shahak, S. Tsaroom and J. Blum, J. Org.
Chem., 1978, 43, 4271; J. Legters, L. Thijs and B. Zwanenburg,
Tetrahedron Lett., 1989, 30, 4881.
-
Y. Hu, A. Harada and S. Takahashi, Synth. Commun. , 1988, 1607.
-
J. Auge and S. David, Tetrahedron Lett., 1983, 24, 4009.
-
I. Coldham, A.J. Collis, R.J. Mould and D.E. Robinson, Synthesis,
1995, 1147.
-
A. Osuka and H. Suzuki, Tetrahedron Lett., 1983, 24, 5109.
{kind=link}