
 When Jacobsen introduced the salen-managanese catalysted epoxidation,
he proposed an orientation of the olefin parallel to the salen plane, and
perpendicular to the metal-oxygen bond.[15] This 'side-on' approach
of the olefin, was postulated by analogy to the iron epoxidation mechanism
proposed by Groves.[16]
When Jacobsen introduced the salen-managanese catalysted epoxidation,
he proposed an orientation of the olefin parallel to the salen plane, and
perpendicular to the metal-oxygen bond.[15] This 'side-on' approach
of the olefin, was postulated by analogy to the iron epoxidation mechanism
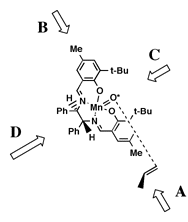
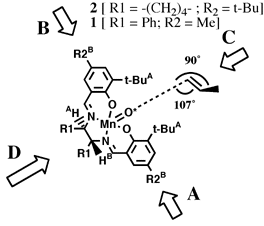
proposed by Groves.[16]Jacobsen hypothesized that the olefin would approach the oxidized catalyst over the aromatic salen ring farthest from the raised phenyl group. This approach ('A' in Figure 5), allowed the olefin to avoid the bulky tert-butyl groups blocking approach 'C', and the raised phenyl group hindering approach from either 'B' or 'D'. Jacobsen further suggested that the olefin would be orientated in such a way to place substituents on the olefin as far from the tert-butyl groups as possible, and pointed away from the salen ring - resulting in the observed stereoselection.
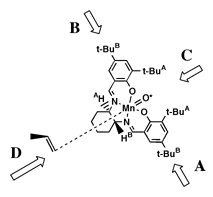
This hypothesis effectively predicted the selectivity of the diphenyl catalyst. However, when Jacobsen introduced his second--and more selective--catalyst the observed selectivities were exactly the opposite predicted by this model. Jacobsen suggested a different approach for the cycloheyxl catalyst (1) catalyst. As tert-butyl groups were now added to the ortho position of the salen aromatic rings, Jacobsen proposed that approach from both faces 'A' and 'B' (Figure 6) would be now be blocked. As approach 'C' would also be blocked by tert-butyl groups, it seemed likely that olefin must approach from direction 'D.' Stereoselection could then be interpreted to result from the olefin attempting to orient the bulky substituents away from the salen plane and as far from the cis-axial hydrogen as possible.
While each of these mechanisms predicts the correct sense of stereoselection in the respective catalysts, we propose an alternative theory which allows a single mechanism to explain the selectivity in all salen-manganese epoxidation catalysts.
Jacobsen has emphasized the similarity between salen based catalysts and their porphyrin based predecessors. In his first paper on this reaction, he presented a crystal structure of a salen complex di-ligated with acetone and commented that the salen 'tetradentate ligand adopts a near planar geometry.'[17] However, salen ligands have a fundamental difference from porphyrin ligands: while the latter is composed exclusively of sp2 atoms, salen ligands have exactly two sp3 atoms which link the remaining sp2 atoms. The preference of a gauche rather than an eclipsed conformation of this linkage causes the salen system to twist, like two planes leaning on a third - each touching the ground on an opposite side. In preliminary modelling investigations we observed a twisting of the two salen aromatic rings away from planarity caused by this linkage and postulated that this twisting might be the source of chiral induction in the catalyst.
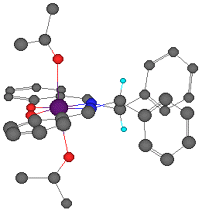
The linking sp3 centres impart a significant twist to the aromatic planes, as shown in Figure 7. Figure 7 is a reproduction of Jacobsen's crystal structure (recovered from the Cambridge Structural Database) but viewed from a different perspective than it was originally presented. The observed twist is pronounced, resulting in an angle of 12o between the aromatic rings of the salen ligand.
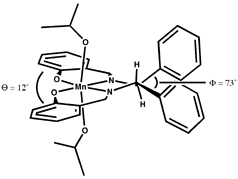
The sense of the twisting is consistent with the enantiomer of the epoxide formed. In all enantioselective salen catalysts the linking carbons are stereogenic, and act in concert to impart a favoured twist to the ligand. In Jacobsen's first catalyst (2) the linking carbons bear phenyl substituents which must either both be axial or both equatorial relative to the metallacycle, resulting in a strong preference for the di-equatorial conformation. The linking carbons in Jacobsen's cyclohexyl catalyst (1) are embedded in a six-membered ring which obviously demands gauche equatorial positions, locking the two aromatic halves of the salen complex into a preferred twist.
As seen from approach C (Figure 8), both (R,R) 1 and (R,R) 2 have the 'left' salen aromatic ring tilted below the Mn-N-N plane, and the salen aromatic ring on the 'right' tilted above it (defined here as a counterclockwise twist). Both demonstrate a preference for formation of the same epoxide enantiomer.
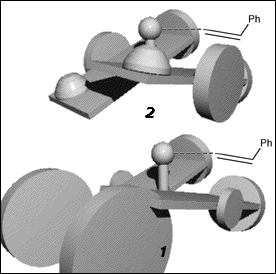
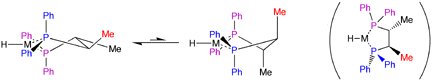
We postulate that the stereogenic centres on the linking carbons transmit their chirality through the body of the catalyst to the remote C face, where reaction with the olefin--and thus the stereoselection--occurs. This kind of remote stereocontrol is precedented by Bosnich,[18] who studied how chirotopic substitution of a diphos ligand resulted in enantioselective hydrogenation (illustrated in Figure 9).
Accepting that the epoxidation occurs by a stepwise radical addition of the activated oxygen (O*) as Jacobsen has proposed, the transition state essential for determining the degree of enantioselectivity will lie along the reaction coordinate of the first carbon oxygen bond forming step. By analogy to carbon radical additions to double bonds, we expect the forming bond to have a 107o angle relative to the plane of the olefin and a 90o dihedral angle relative to the substituent on the olefin while perpendicular to the O*-Mn bond as illustrated in Figure 8.
In monosubstituted olefins, the catalyst will add to the olefin to produce the more stabilized radical.
We postulate that the olefin carbon not forming the initial bond will move as far from the raised But substituent as possible, adopting the orientation shown in Figure 8. The substituent moves as far from the salen ligand as possible. This model is also consistent with the observed selectivities in di- and tri-substituted olefins. The olefin tries first to avoid the raised But group and secondly to keep its substituents as far from the salen ligand as possible.
Each of the 18 manganese salen complexes in the Cambridge Structural Database indicate the same twisting to varying degrees.
In all enantioselective salen maganese complexes of which we are are, this single model predicts the correct sense of stereoinduction.
