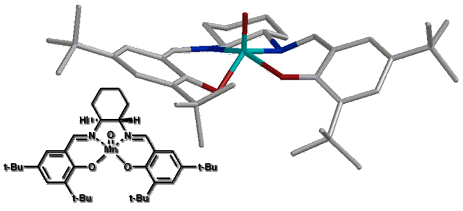
For our initial study we created a molecular mechanics forcefield to describe the ground state of salen manganese catalysts based on X-ray crystal data. This preliminary forcefield was modified by addition of transition state bond lengths for the olefin-oxygen bond distance and the olefin oxo-manganese dihedral angles based on quantum mechanic calculations. This allowed us to optimize transition state geometries for various olefins at different approaches to the optimized oxo-catalyst structure (Figure 10). As this forcefield is based only on ground state crystal structure data and simple, low level ab initio calculations, the resulting geometries should only be interpreted to suggest trends in the reaction surface. A more reliable forcefield - based on high level ab initio determined structural data - is still in development.
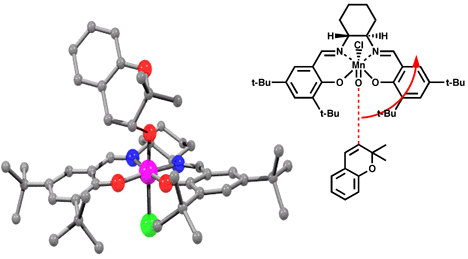
Models of olefins approaching both of Jacobsen's catalysts at various approaches to the catalyst were prepared. These models were distinguished by the dihedral angle between the forming olefin-oxygen bond, the oxygen manganese bond, and an imaginary line from the manganese to the mid-point between the two salen oxygens. Varying this locked dihedral angle in ten degree increments before optimizing each geometry resulted in the energy profiles shown in Figure 11 and 12. Both the favoured and disfavoured diastereomeric transition states were examined. A clear preference was shown for approach of the olefin from the 'C' face.
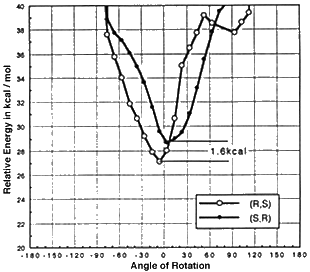
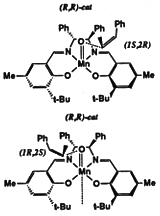
Figure 11 Rotational profile of diphenyl catalyst
These calculations indicated a preference for the major observed epoxide for the diphenyl catalyst (by 1.6 kcal mol-1 1 cal = 4.184 J), and a greater preference (3.0 kcal mol-1) for the major observed epoxide with the more selective cyclohexyl catalyst.
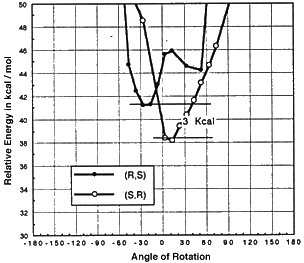
Figure 12 Rotational profile of cyclohexyl catalyst
Further development of this forcefield requires a more accurate description of the transition states for epoxidation.
Ab initio ground state calculations of manganese oxo complexes have consistently indicated a triplet ground state is preferred. The most reliable ab initio method we have explored has been density functional calculations with the dzvp gbs.[] Unfortunately the calculation of even simplified analogues of the catalyst has taken weeks on an IBM RS6000 (Figure 13). We have found that the semi-empirical method PM3(tm) can generate very similar structures (with bond length variations of only 0.05 Å from dzvp gbs structures) in only 4 h.
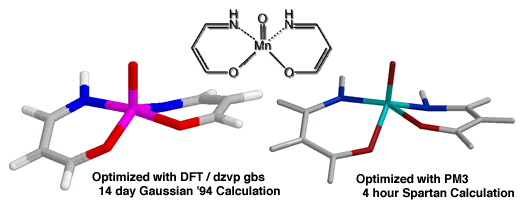
The computationaly more economical PM3(tm) methods have been employed to solve the ground state structure of both the diphenyl and cyclohexyl catalysts (Figure 14) - both of which demonstrate a pronounced twisting of the salen aromatic rings. Further studies of this system are underway, including attempts to solve the transition state of epoxidation with PM3(tm) methods and to determine key interaction parameters with DFT/dzvp gbs.