Previously, I explored deviation from ideal tetrahedral arrangements of four carbon ligands around a central (sp3) carbon using crystal structures. Now it is the turn of digonal (sp1) and trigonal (sp2) carbons.
Firstly, the digonal C≡C case. Attached to each carbon of the C≡C unit are two saturated carbon ligands; this to prevent conjugation from influencing our result.
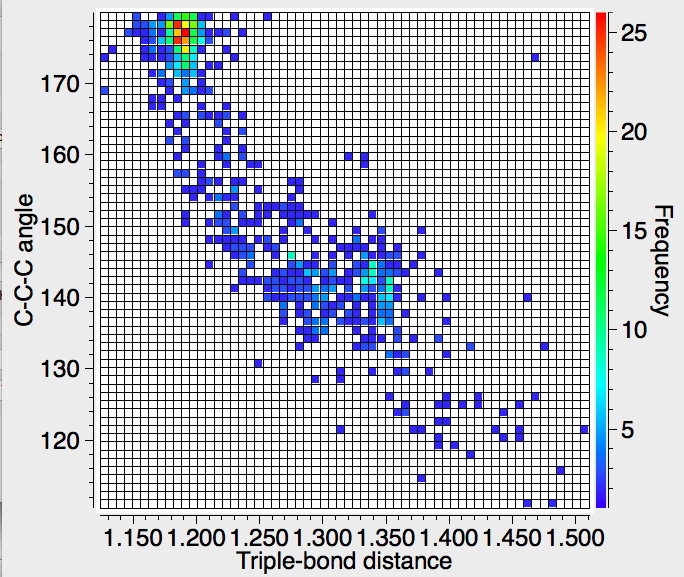
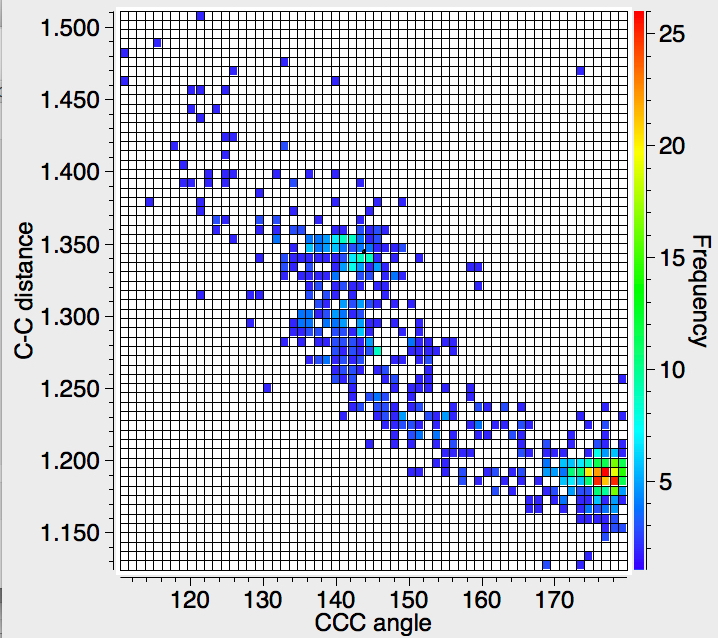
The result of a search (R-factor < 5%, no errors, no disorder) shows the hotspot at the expected ~180°, but then a fascinating curve as the angle subtended at the digonal carbon angle decreases down to ~110°, with the C≡C bond length gradually increasing. This apparently non-linear behaviour would be interesting to replicate using quantum mechanics.‡
Next, the trigonal case. Again, the substituents are 4-coordinate carbons to prevent complicating conjugations.
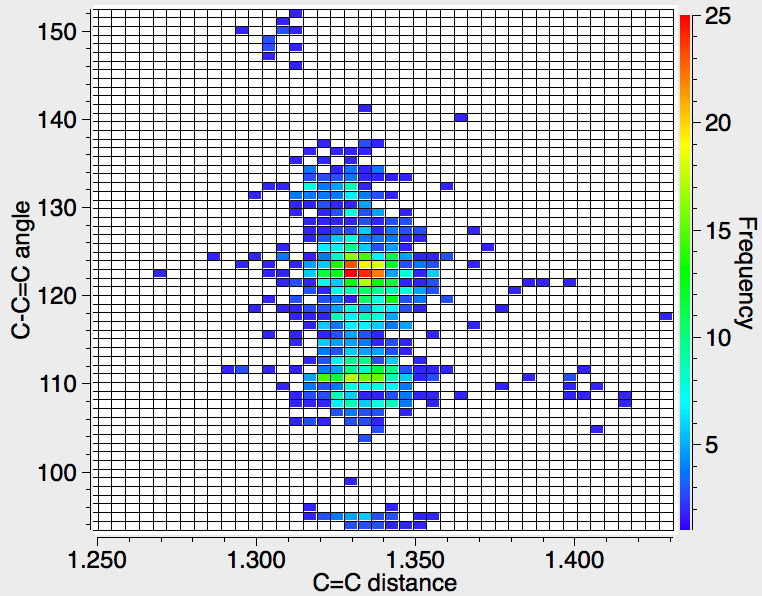
A plot of the C=C distance vs the C-C=C angle brings a surprise. There are four clusters centered at angles of ~132°, 123°, 110° and 94° (cyclobutenes) and a small cluster at ~150°. The C=C distance stays constant at around 1.335Å or shorter, a clear difference with the sp-case. There is perhaps a small outlier collection where the angle is ~108° and the distance ~1.4Å.
This plots the dihedral angle subtended at one of the trigonal carbon atoms and measures how non-planar that atom is. There is again no real evidence that the C=C bond length changes as the trigonal centre becomes bent.
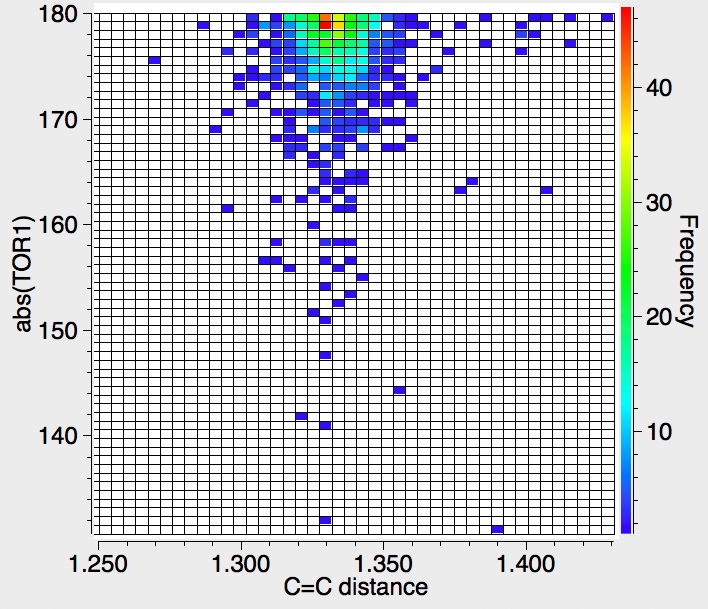
This dihedral angle measures the twist about the C=C bond; up to about 30° is tolerated, but again there is no clear indication of a systematic change in the C=C length.
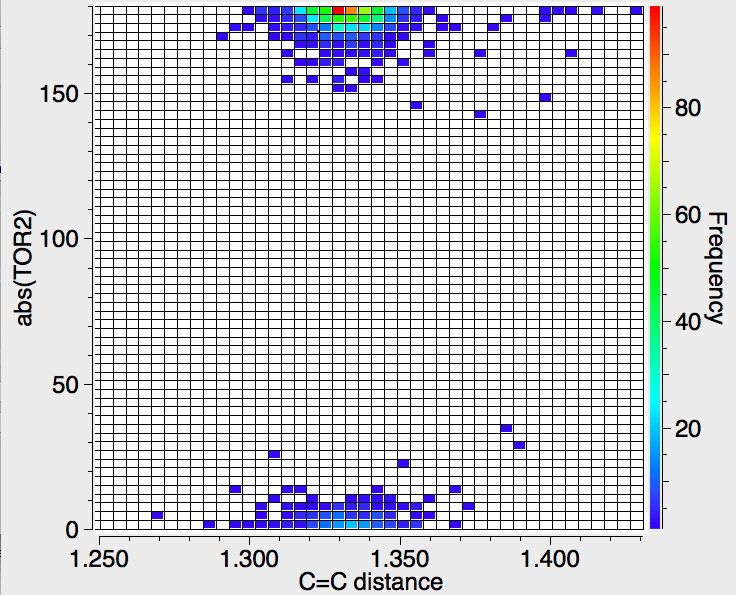
These analyses reveal general trends on bond lengths induced by distorting the normal coordination around trigonal and digonal carbon atoms. It is only the start of the story of course, since there are plenty of isolated outliers that really should be explored; some may be simply due to undetected crystallographic errors, whilst with others there may lurk interesting or even new chemical phenomena.
‡Below, the crystal structure result (with the axes transposed) is compared to a closed shell single reference ωB97XD/6-311+G(2df) calculation. Whilst the trend is replicated, it is not quantitative. This is probably because many of the crystal structures are perturbed by other effects, most probably by coordination of a metal and hence back-donation of π-electrons into vacant metal orbitals. The CSD indexing of the structures however retains the C≡C bond notation, even though the bond is no longer truly a triple one. This reinforces the observation I made in the previous post that when searching the CSD, one can stipulate a bond type to constrain the search. But that bond type may be purely nominal and bear little resemblance to the actual electronic structure of the species. There are other issues; the wave function was constrained to closed shell single determinant. At low angles, the calculation itself is probably not accurate (as can be seen from a kink in the plot, indicating instability).

Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
Tags: chemical phenomena