The title of this post summarises the contents of a new molecular database: www.molecularspace.org[1] and I picked up on it by following the post by Jan Jensen at www.compchemhighlights.org (a wonderful overlay journal that tracks recent interesting articles). The molecularspace project more formally is called “The Harvard Clean Energy Project: Large-scale computational screening and design of organic photovoltaics on the world community grid“. It reminds of a 2005 project by Peter Murray-Rust et al at the same sort of concept[2] (the World-Wide-Molecular-Matrix, or WWMM[3]), although the new scale is certainly impressive. Here I report my initial experiences looking through molecularspace.org
The 150,000,000 calculations are released under the the CC-BY license, which is an encouraging (open) start. One does need however to login to the site, which I was able to do using my Google credentials. Shown below is a screenshot of a typical result in a search (of Power conversion efficiency in my case).
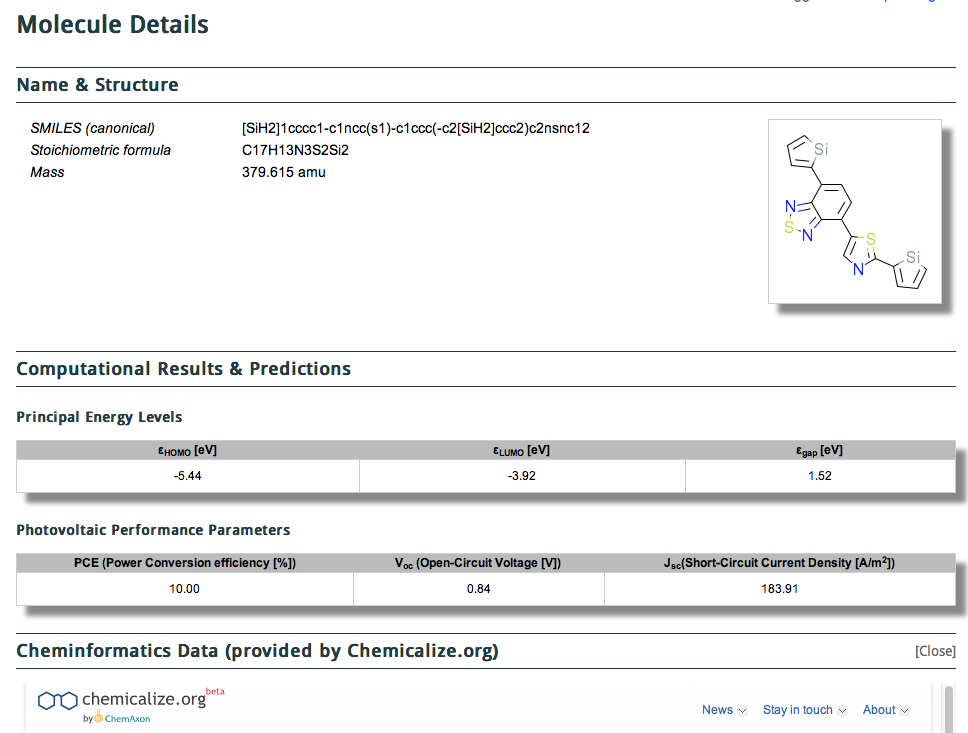
It comes in two parts, the first being the structure (given as a SMILES and 2D layout) with the principle predicted energy levels and predicted photovoltaic performance listed below that. This is then followed by what might be called an annotation with further computed/predicted properties using the algorithms applied by Chemicalize.org. This idea that a data set could accrete via semantically powerful annotations using other tools was also very much part of the concept of the WWMM (the matrix had at its heart a molecule in one dimension and a property, measured or computed in the other. The matrix is of course very sparse, which is why it needs annotation!).
It was at this point however that I started to wonder how I might add other annotations, based perhaps on other types of calculations. But thus far at least, I have not found any trace of something which I could immediately use for my own calculation; 3D coordinates specifically. Thus, the HOMO-LUMO energy gap is the key property which makes molecularspace unique and valuable (to someone working in the field of photovoltaics). But HOMO/LUMO gaps can be calculated in many different ways, and it can always be valuable to calibrate/validate the reported values against other methods. Perhaps if I continue to look, I might find these 3D coordinates (which, for 2,300,000 molecules would be a very valuable resource). Certainly for example, should I wish to do so, I could not at the moment readily replicate the calculation for any specific entry on the molecularspace site (which can be regarded as an essential component of scientific validation). When I use the first person, I mean of course either myself as a human or a software agent acting on my behalf (the latter having the endurance to repeat its procedures millions of times if necessary).
The reader of this blog may have noticed that whenever I report a calculation here, I like to cite its doi (more formally its handle), which links to a digital repository. In my case, the repository certainly carries the 3D coordinates, and also the full wavefunction provided if the reader wishes other properties to be derived from it.‡ Now if molecularspace is able to provide that in the fullness of time, it truly would be an impressive resource.
But the important take-home message from molecularspace is that archiving (under a CC-BY license) the “big” data from any given research in a manner which makes it readily re-usable by others (perhaps from quite different fields of science) is now an essential requisite of doing science. And it is really nice to see good examples of this in practice!
‡ Generally, the calculations I perform for this blog are published in a DSpace repository (the original one, started in 2006[4]), and more recently in Chempound (a project by Peter Murray-Rust and colleagues which emerged out of the WWMM experiments) as well as Figshare[5]. The first and the third assign unique handles (i.e. a doi) to the data; chempound does not (and neither does molecularspace).
Author
References
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R.S. Sánchez-Carrera, A. Gold-Parker, L. Vogt, A.M. Brockway, and A. Aspuru-Guzik, "The Harvard Clean Energy Project: Large-Scale Computational Screening and Design of Organic Photovoltaics on the World Community Grid", The Journal of Physical Chemistry Letters, vol. 2, pp. 2241-2251, 2011. https://doi.org/10.1021/jz200866s
- P. Murray-Rust, H.S. Rzepa, J.J.P. Stewart, and Y. Zhang, "A global resource for computational chemistry", Journal of Molecular Modeling, vol. 11, pp. 532-541, 2005. https://doi.org/10.1007/s00894-005-0278-1
- P. Murray-Rust, S.E. Adams, J. Downing, J.A. Townsend, and Y. Zhang, "The semantic architecture of the World-Wide Molecular Matrix (WWMM)", Journal of Cheminformatics, vol. 3, 2011. https://doi.org/10.1186/1758-2946-3-42
- J. Downing, P. Murray-Rust, A.P. Tonge, P. Morgan, H.S. Rzepa, F. Cotterill, N. Day, and M.J. Harvey, "SPECTRa: The Deposition and Validation of Primary Chemistry Research Data in Digital Repositories", Journal of Chemical Information and Modeling, vol. 48, pp. 1571-1581, 2008. https://doi.org/10.1021/ci7004737
- H.S. Rzepa, "Gaussian Job Archive for CLi6", 2013. https://doi.org/10.6084/m9.figshare.739310
Tags: energy gap, energy levels, Google, Harvard, Jan Jensen, molecularspace site, opendata, Peter Murray-Rust, software agent acting, www.compchemhighlights.org, www.molecularspace.org
Hi,
Interesting read. I found that you can in fact download the structures (albeit without hydrogens). When you have located a structure, in the bottom there is a section labelled “cheminformatics data” – right below that you hit “manage calculations” and under “geometry” you click “geometry” – there is a rendered 3D structure. Double click it to get a marvin interface in java up and here you can save to whatever format you want. cumbersome. Perhaps it just translates the smiles string into a 3D structure.
You raise an interesting point about the provenance of data. Such provenance, or meta-data, tells the prospective re-user of the data how it was generated (and hence the context of how it can be re-used).
In this example, the database exposes a DFT calculation of the HOMO-LUMO energies, and then following this is cheminformatics data (provided by Chemicalize.org).
Immediately, we become unsure whether the available data in this section (such as the geometry) is directly inherited from the preceding DFT calculation, or whether it has been changed by some implicit (undeclared) algorithm. Can we trust this geometry? Is its provenance provided by the DFT method, or by Chemicalize?
One might try to track this down (from the Chemicalize site). As a human, I gave this a quick attempt, but did not immediately track down for certain how “3D” structures might be generated there. It is probably done using a simple layout algorithm since the .sdf format file provided using “download results” has zeros for the z-coordinate (for a manifestly non-planar structure). I was able to get this far because I am human, but a machine (software agent acting on my behalf) would not have been able to do this.
So at this stage, it is probable that the geometry data provided on the molecularspace site has a provenance from Chemicalize (a separate organisation), and is not related to the coordinates used for the DFT calculation. We might also conclude that this data is not suitable for eg alternative calculation of HOMO-LUMO gaps. Because the third z-coordinate in the Chemicalize data is set to zero, we may also infer that stereochemical information (about eg stereoisomers) is also lacking from this data.
I think this shows how necessary it is to fret about the provenance of data, and its suitability for any particular re-use. Particularly if one wanted to say re-use a significant fraction of the data using an automated mechanism.
The reference to Figshare[4] does not seem to work. 🙁
In fact, it is an interesting idea that you “publish” the Supporting Information in a separate repository, which I was interested in the Figshare.
(I found it though: http://figshare.com)
Will have a look at it. Thanks!
The figshare link was broken by an extraneous character. Try again.
Regarding the SI in a separate repository; virtually all conventional SI is currently not published in the “correct manner”. In other words, it has to have proper meta-data, including the (ORCID?) identity of the depositor, the deposition date, and built in support for a number of repository protocols (including SWORD, which allows one to transfer data from one repository to another). Most importantly, a digital repository can have elaborate “quality control” checks running prior to deposition. Conventional SI has no mandatory structures, metadata, or QA. Often, it can be just a TIFF file with minimal caption. By assigning the SI its own DOI, one might be assured that many of these requirements are in place (as one is assured of the provenance of an article carrying a DOI).
More on Figshare. They have published an API (application programmer interface) which allows one to construct an interface for deposition. We are building such an interface for chemistry (based on php scripts), which will serve the purpose of capturing the provenance and other metadata of a dataset (we did this first for an older digital repository, DSpace, a few years ago).
Hi Henry,
Pertaining to this, you might be interested in some new CML functionality within NWCHEM’s output; http://dx.doi.org/10.1186/1758-2946-5-25 . Imagine the re-usability value if this experiment had been done with the output described in this paper and then deposited to something like ChemPound.
Thanks Mark. The article you cite also includes Avogadro as the front end for the NWCHEM program. In one regard at least, Avogadro breaks new ground; it does not have a native file format, but instead uses CML for this purpose. Why is this ground breaking? Because not only does it capture the semantics, it does so in an open and exposed manner, and one in which other programs and processes have full unencumbered access to. Contrast this with older approaches using proprietary formats which have to be constantly reverse-engineered to gain access to the semantics.
So I fully agree how wonderful it would have been if molecularspace had adopted this approach. I again repeat that the only accessible semantics that appear to be available there are those added by Chemicalize. And I presume this is there because of some bilateral agreement between the two organisations. Much better to do it in a manner which would allow anyone to annotate with added semantics, rather than being limited to bilateral agreements.