The title here is taken from a presentation made by Ian Bruno from CCDC at the recent conference on Open Science. It also addresses the theme here of the issues that might arise in assigning identifiers for any given molecule.
The structure was represented as shown[1] by the original authors, in which the bonding from S to Sn is indicated with both solid lines (a bond) and dotted lines (an “interaction”).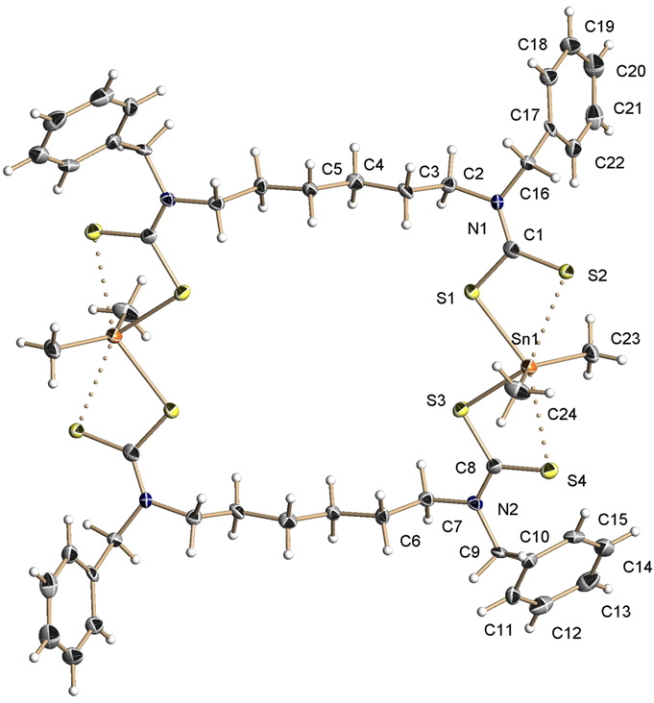
Why would this matter? Well, to enable any entry in the Cambridge structure database as findable (the F of FAIR) it has to be given a unique identifier. There are in general three such identifiers assigned by the CCDC:
- The Refcode, in this case XONHIS. These six or seven letter codes are historically the oldest, and started off at least with an attempt if possible to assign some semantic inference from the name, even if only occasionally.
- The CCDC deposition number, in this case 650011. This is the number that an author will receive immediately upon deposition, and you often find these identifiers quoted in supporting information files
- The DOI (digital object identifier), in this case 10.5517/ccptd3z, which can be used to view the structure even if access to the full CSD is not available to the user. In that sense, the DOI is the FAIRest of the first three of these identifiers.
- However, CCDC reported that they are considering adding a 4th very common identifier, based on the InChI (International chemical identifier), which comes as a full string and with the structure of the molecule at least in part inferrable from it, together with a shortened (almost) unique string which has the advantage of being “Googlable”. Both are helpfully FAIR.
It is this 4th identifier that is at issue here. InChIs are derived from atom connection tables; you need to define all bonds present in the molecule. And it is here that the dotted “bond”/”interaction” above becomes a problem. This is the representation shown in the CSD database, which reveals that all the Sn…S interactions are classified as “bonds”, along with some creative(!) representations of the C…S bonds.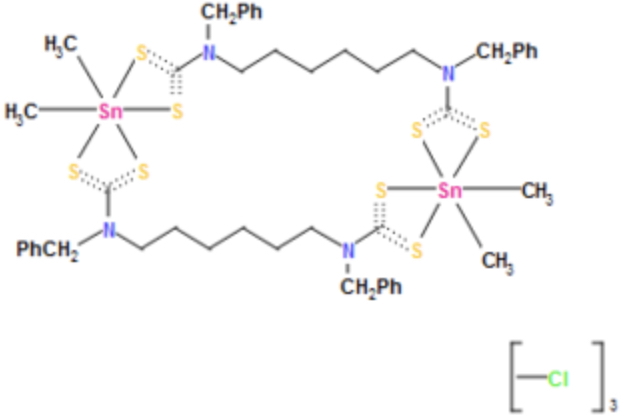
So the InChI will very much depend on whether all the Sn…S contacts are termed as bonds or as interactions. To help clarify that, it is useful to show the typical range of lengths of such contacts. Below is a simple search for all Sn and S systems where the pair are either close in space (< 3.5Å) or have a bond specified between the two atoms.
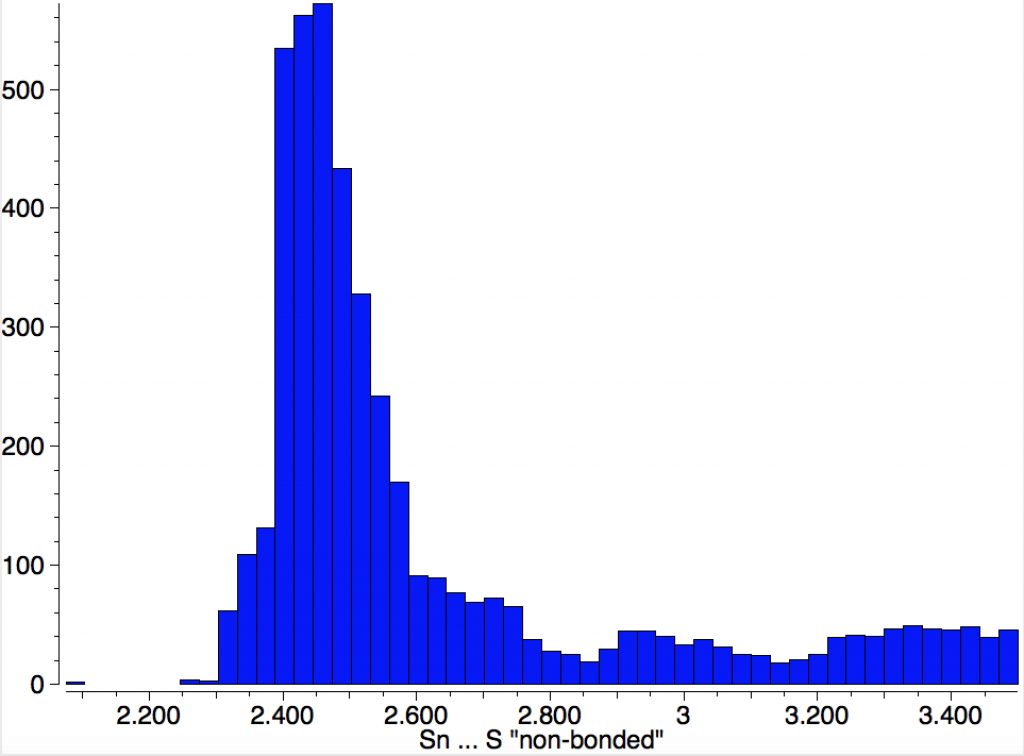
The main cluster occurs at ~2.5Å, but there is some evidence of a second peak at about 3.0Å. The third distribution up to 3.5Å is probably a continuum of very weak dispersion interaction, which most molecules exhibit. The values for XONHIS are 2.521 and 2.996Å, which match the two clusters above.
So perhaps a quantum calculation can shed some light (DOI: 10.14469/hpc/2593)? The values on the right are the optimised bond lengths which are pretty similar to the crystal structure. On the left are the calculated Wiberg bond orders (B3LYP+D3BJ/Def2-TZVPP/chloroform calculation). These reveal both “bonds” have an order less than 1. The value of ~0.6 is probably not contentious, but it does graphically show that when a compound is indexed as having a “single bond” between two atoms, the quantitative bond order may be substantially less. What however would one make of a bond order of 0.214? Should it be classified as a bond, albeit a much weaker one than normal? Or should it instead simply be a rather strong “interaction” which is not classified as a bond? And perhaps one should have in mind the question “how sensitive is this result to the quantum mechanical procedure used?”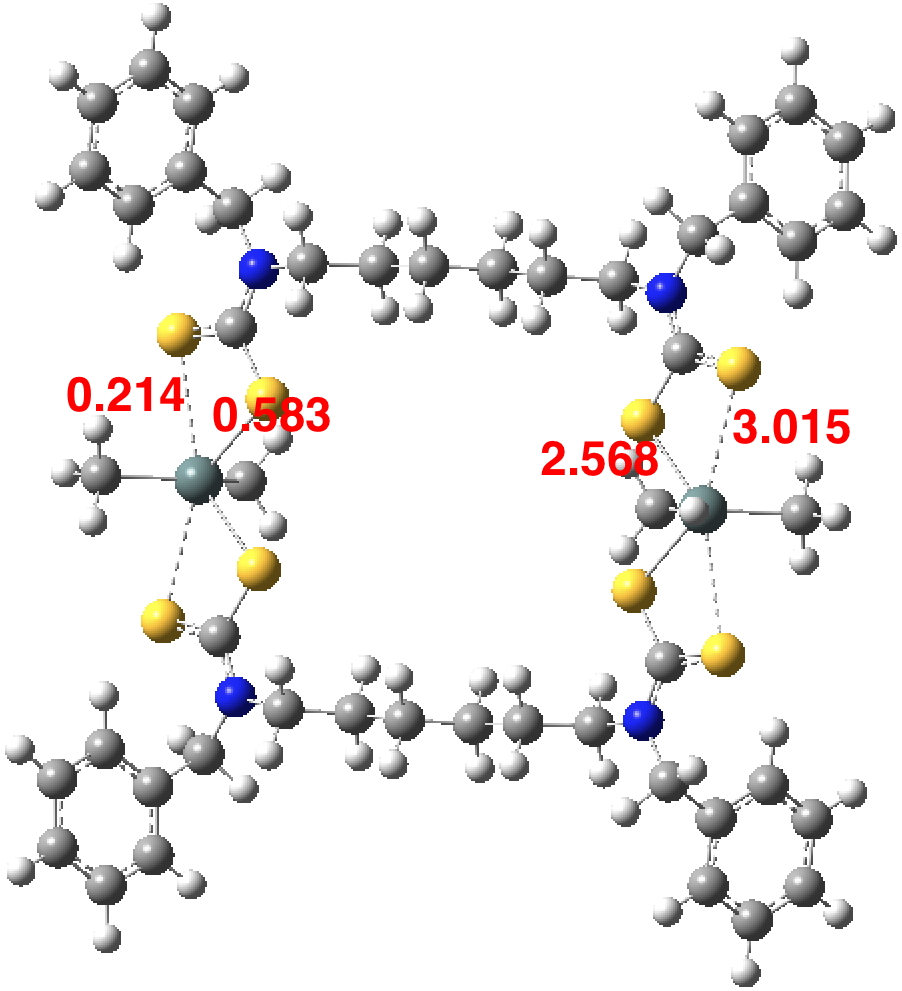
Why does this distinction matter? Well, the InChI algorithm is based on simple connectivity; are two atoms connected by a bond or not? There are no nuances here. At the moment, this decision can be made by an algorithm based on the distance between any atom pair (whether computed or measured), but more often I suspect it derives from a “molfile” which is often derived from a human-drawn representation using a structure drawing program. It does rather boil down to the individual preferences of the human drawing the molecule. Due in part to such uncertainties, it was estimated that only 22% of structures in the CSD can be used to generate a reliable InChI. Hydrogen bonds are almost always classified as non-bonds, which means their presence is rarely systematically flagged during the indexing of the structures. Organometallics often pose some of the greatest representational problems (there are many others).
I will end by observing another class of structure that I deal with, “reaction transition states”. As you might imagine these forms are full of pairs of atoms with ambiguous bond lengths and hence connectivity. We currently have no truly reliable method for assigning useful identifiers to them. So lots of challenges for the future then!
Author
References
- R. Reyes-Martínez, R. Mejia-Huicochea, J.A. Guerrero-Alvarez, H. Höpfl, and H. Tlahuext, "Synthesis, heteronuclear NMR and X-ray crystallographic studies of two dinuclear diorganotin(IV) dithiocarbamate macrocycles", Arkivoc, vol. 2008, pp. 19-30, 2007. https://doi.org/10.3998/ark.5550190.0009.503
Tags: author, Bruno, chemical identifier, Digital Object Identifier, Ian Bruno, Identifier, InChI algorithm
So – why not define a “standard connectivity metric method”, by which interactions can be reproducibly classified?
Say:
1) Perform a calculation on the system at a specified level of theory (say B3LYP), with a specified basis (say 6-31G**), with a specified (open source/freely-available, of course!) quantum software.
2) Calculate Wiberg (or some other specified standard) bond order indices for the entire system, for all pairs of atoms separated by smaller than some large threshold distance, say 5 Angstroms.
3a) If feasible, store the entire library of bond order indices themselves as the fingerprint for the system.
3b) If not feasible, define thresholded ranges for the bond order index that classify the pairwise interactions into categories comprehensible to, e.g., InChI.
1,2,3b would at least go a long way toward removing user-specific variations in the connectivity definitions; 1,2,3a would result in a substantial body of bond order information being deposed along with the crystal structure.
I’m sure it’s not actually this simple to come up with something robust (high-spin radical systems and species with large static correlations come to mind as spoilers), but surely there’s *something* that would cover a majority of cases?
One of the unique features of InChI is that it can be computed extremely rapidly (at virtually no cost) by anyone who acquires the executable code, in any context and in very high throughput (i.e. scanning whole databases). Adding the requirement that an InChI must be derived using a quantum mechanical procedure (even assuming that a consensus could be agreed about the Hamiltonian/basis etc) would make it an expensive identifier to compute. Remember there are some very large molecules for which InChIs are nowadays computed. And those for the RInChI (reactions), MInChI (mixtures) and polymers may be even larger. Since reliable and general quantum mechanics scales at best as ~N3, the size of the molecule would rapidly cause problems.
If a solution is possible, I think it will require an entirely new approach.