|
|
|
kCal.mol-1
| H°f(reactants) | H°f(T.S.) | H°f(products) | Reaction Enthalpy | Activation Energy | ||
|---|---|---|---|---|---|---|---|
| Semi-Empirical MOPAC 93 | PM3 | endo | 17.76 | 41.81 | -4.73 | -22.48 | 24.05 |
| exo | 38.65 | -7.88 | -25.63 | 20.90 | |||
| AM1 | endo | 55.33 | 73.44 | 25.80 | -29.53 | 18.11 | |
| exo | 70.62 | 23.53 | -31.80 | 15.29 | |||
| Molecular Mechanics (CAChe) | MM2 | endo | -27.50 | 8.80 | -13.51 | 13.99 | 36.30 |
| exo | 8.19 | -11.55 | 15.94 | 35.69 | |||
kCal.mol-1 |
Reaction Enthalpy | Activation Energy | |
|---|---|---|---|
| Experimental | endo | -27.0 | 26.3 |
| exo | -29.2 | 25.1 | |
Table 2.2 : Experimental results
With the MM2 Force Field we get an endo thermodynamic selectivity. Experience has shown that is is not, and we will have to keep this correction in mind to compare with the calculation in the presence of a catalyst. The value we get for the transition state is not very useful because we cannot describe the hybridization of the different atoms. For example, the oxygen atom on the diene is sp2 in the reactants and sp3 in the product. We cannot model it in the transition state in a way that can be treated correctly in a molecular mechanic calculation. Furthermore, the transition state is not a minimum of energy. The algorithms that are used to find the geometry of the ground state do not converge rapidly to the transition state.
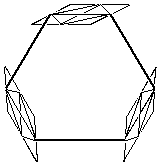 The planes represent the porphyrins and the bold lines represent the links between them. |
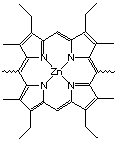 The Zinc-porphyrin |  Links used in Catalyst 1 |
 Links used in Catalyst 2 |
Figure 2.2 : Catalysts 1 and 2
kCal.mol-1 |
H°f(reactants) | H°f(T.S.) | H°f(products) | Reaction Enthalpy | Activation Energy | |
|---|---|---|---|---|---|---|
| MM2 | endo | -129.00 | -40.72 | -98.42 | 30.58 | 88.28 |
| exo | -90.69 | -108.82 | 20.18 | 38.51 | ||
| endo adduct | exo adduct | |||
|---|---|---|---|---|
| Mopac 93 PM3 | H°fkCal.mol-1
| Gradient Norm | H°fkCal.mol-1
| Gradient Norm |
| 850.1 | 8.47 | 842.0 | 6.74 | |
Table 2.4 : Semi-Empirical (PM3) results
kCal.mol-1 |
Reaction Enthalpy | Activation Energy | |
|---|---|---|---|
| Experiments | endo | -27.7 | 27.3 |
| exo | -28.9 | 24.2 | |
Table 2.5 : Experimental results[11]
kCal.mol-1
| H°f(reactants) | H°f(T.S.) | H°f(products) | Reaction Enthalpy | Activation Energy | |
|---|---|---|---|---|---|---|
| PM3 | endo | 136.02 | 105.23 | -116.21 | 19.82 | 68.08 |
| exo | -90.93 | -117.06 | 18.96 | 43.08 | ||
| kCal.mol-1 | [ Catalyst ] | Catalyst Ground State |
Deformation | [ Substrate ] | Substrate Ground State |
Strain | |
|---|---|---|---|---|---|---|---|
| Catalyst 1 | Reactants | -87.70 | -89.30 | 1.60 | 12.19 | -27.50 | 39.69 |
| Exo Product | -87.86 | 1.44 | 29.12 | -11.56 | 40.68 | ||
| Endo Product | -87.48 | 1.82 | 40.80 | -13.51 | 54.31 | ||
| Catalyst 2 | Reactants | -85.99 | -90.04 | 4.05 | 25.33 | -27.50 | 52.83 |
| Exo Product | -79.22 | 10.82 | 28.66 | -11.56 | 40.22 | ||
| Endo Product | -82.60 | 7.44 | 29.14 | -13.51 | 42.65 | ||
The MM2 force field does not describe the dative bond between nitrogen and zinc. We require CAChe Mechanics to estimate the missing parameters : using the augmented option, the calculation was extended to all elements by applying simple empirical rules to estimate parameters for elements not in the MM2 set. This explains why the strain energies for the substrates are very high. We are only interested in the difference between these numerical values. For example, it is obvious that the endo adduct is much more strained than the exo adduct (about 14 kCal/mol). We note that for catalyst 1, the deformation is not important (less than 2 kCal/mol) whereas with catalyst 2, the smaller one, it is more significant. Catalyst 2 is probably too small for catalysing either the endo or the exo additions.
The same experiment was carried out using the PM3 semi-empirical method for catalyst 1. The single point energies of the two parts of each catalyst+substrate complex are shown in Table 2.8 :
| kCal.mol-1 | [ Catalyst ] | Catalyst Ground State |
Deformation | [ Substrate ] | Substrate Ground State |
Strain |
|---|---|---|---|---|---|---|
| Exo Product | 867.5 | 849.7 | 17.8 | -6.2 | -7.9 | 0.8 |
| Endo Product | 865.3 | 15.6 | 5.2 | -4.7 | 9.9 |
We first have to check the validity of this analysis : the energy of the two dative bonds is
These results partially invalidate the deduction we could have made with the MM2 experiments. We find here that the exo adduct is practically unstrained, whereas there is a strain energy at about 10 kCal/mol for the endo adduct. This confirms that the porphyrin trimer has exactly the right size to bind the exo product. The deformation of the catalyst is more important than has been found with mechanics calculations, but it remains very small in comparison with the size of the catalyst: the deformation is spread out over the 285 atoms and the 315 bonds.
Semi-empirical calculations are a solution to this problem, but they require so much time that we have to be sure of the interest of the computation. In the near future, with the increase in the capacities of the computers, it will probably be possible to carry out these calculations within a week or less. Another solution may lie in running Mozyme. Mozyme is a semi-empirical quantum chemical program for the study of large systems. The methods in Mozyme are similar to those in Mopac. The fundamental difference is that, whereas MOPAC uses matrix algebra for the solution of the self-consistent field equations, Mozyme uses localized molecular orbitals (Lewis structures).
The idea of trying to model new catalysts can be applied to other classes of reactions. We could, for example, try to build an asymmetric trimer that could yield the endo product. We have not dealt with entropies. We have already seen that the entropy is not important when we compare two similar reactions like endo and exo additions. If we want to prove that a catalyst accelerates a reaction, we will have to take it into account, because catalysts often act as "entropic-traps" and freeze degrees of freedom. Unfortunately, the estimation of the entropy of formation for each molecule requires much more CPU-time than the heat of formation. That explains why we cannot verify the importance of entropy factors in this system.
| Previous | Table of Contents | Next |