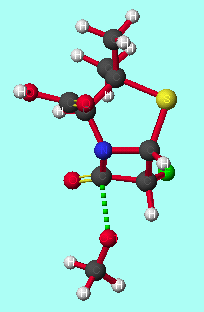
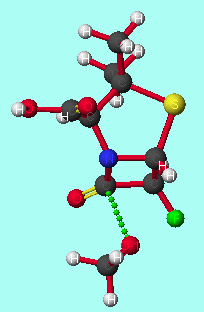
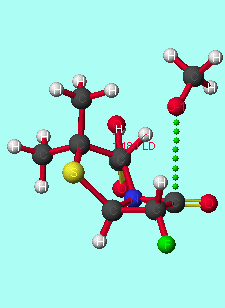
The model reaction between alpha-halopenicillins and a methoxide anion (serine analogue) was studied at the PM3 or AM1 levels. Such methods have been successfully applied to stereoelectronic rationalisation of several other reactions[8]. The transition states were modelled in the gas phase, as it is expected that a substrate bound to the enzyme will be essentially desolvated at the active site[9]. Stereoelectronic control will be maximised under these conditions, as solvation stabilises the oxy-anion orbital; this increases the energy difference between this orbital and the unoccupied C-F [sigma]* orbital, reducing the perturbation stabilisation.
|
|
|
| | anti attack (convex)
| syn attack (convex)
| anti attack (concave)
| syn attack (concave) | |
|---|
Table 1: Results of modelling reaction of methoxide anion with alpha-halopenicillins
Group Direction Hamiltonian O-C TS Reactant Activation
substituted hydroxyl group used Bond Energy ground Energy
at attack length /kcal state /kcal
alpha-posn in TS/A energy/
kcal
CF3 anti convex PM3 2.261 -310.070 -322.729 12.659
CF3 syn convex PM3 - - -294.390 -
CF3 anti convex AM1 2.580 -288.283 -294.291 6.008
CF3 syn convex AM1 2.388 -280.396 -292.731 12.335
F anti convex PM3 2.291 -194.478 -206.191 11.714
F syn convex PM3 2.763 -182.469 -198.645 16.176
F anti convex AM1 2.594 -178.529 -183.560 5.031
F syn convex AM1 2.671 -169.679 -176.846 7.168
F anti concave PM3 - - -200.237 -
F syn concave PM3 - - -210.127 -
F anti concave AM1 2.774 -181.075 -182.598 1.523
F syn concave AM1 3.274 -186.393 -168.535 (-17.858)
Cl anti convex PM3 2.282 -156.878 -168.873 11.995
Cl syn convex PM3 2.922 -143.669 -160.514 16.845
Cl anti convex AM1 2.580 -139.456 -144.169 4.712
Cl syn convex AM1 3.505 -124.457 - -
Br anti convex PM3 2.280 -144.787 -156.645 11.857
Br syn convex PM3 2.837 -131.307 - 25.338
Br anti convex AM1 2.592 -127.141 -132.165 5.024
Br syn convex AM1 3.775 -111.014 - -
I anti convex PM3 2.279 -122.939 -133.544 10.605
I syn convex PM3 2.965 -108.615 -128.263 19.648
I anti convex AM1 2.617 -114.754 -120.013 5.260
I syn convex AM1 4.046 -99.091 - -
Graph 1
Graph 2
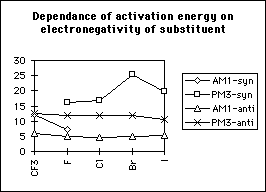
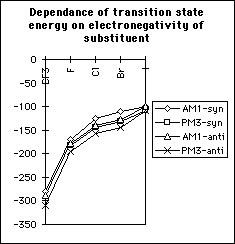
The transition state is of lower energy if the methoxyl group attacks antiperiplanar to the halogen (table 1 and graphs 1&2); the activation energy of the reaction is also lower for antiperiplanar attack than synperiplanar attack. This agrees with the previous modelling and experimental study of alpha-fluoroesters with lipases. This is also seen with other halogen substituted penicillins and the penicillin substituted with the CF3 moiety at the alpha-position. Substituting the penicillin at the alpha-position with substituents of increasing electronegativity results in lower transition state energies; there is no significant correlation between activation energy and elctronegativity. Methoxyl attack from the concave face instead of from the concave face results in higher transition state and activation energies.
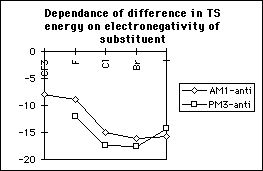
The difference in transition state energies between the two stereoisomers decreases with decreasing electronegativity; however the difference in ground state energies increases with electronegativity and hence the difference in activation energies is independent of electronegativity (table 1, graphs 3,4&5). Therefore there is no correlation between the magnitude of the calculated discrimination of the methoxide anion in favour of antiperiplanar attack and the electronegativity of the substituent at the alpha- position.
Graph 3
Graph 4
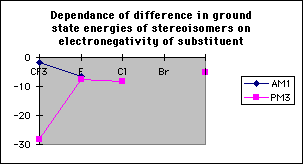
Graph 5
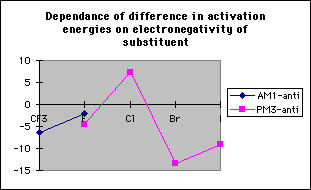
Table 2: Results of frontier molecular orbital calculations for alpha-halopenicillins
Group Direction of Hamiltonian UMO HOMO /UMO Coefficient
substituted hydroxyl group used centred centred on on
at attack on carbonyl carbonyl
alpha-posn carbonyl Energy carbon for
difference UMO
centred on
carbonyl
CF3 anti convex PM3 LUMO+6 9.158 0.124
CF3 syn convex PM3 - - -
CF3 anti convex AM1 - - -
CF3 syn convex AM1 LUMO+5 7.966 0.524
F anti convex PM3 LUMO+5 8.902 0.568
F syn convex PM3 LUMO+5 7.142 0.479
F anti convex AM1 LUMO+5 8.329 0.552
F syn convex AM1 LUMO+4 7.147 0.672
F anti concave PM3 - - -
F syn concave PM3 LUMO +4 8.163 0.688
F anti concave AM1 - - -
F syn concave AM1 LUMO+3 7.527 0.706
Cl anti convex PM3 LUMO+5 9.005 0.301
Cl syn convex PM3 LUMO+4 6.174 0.630
Cl anti convex AM1 LUMO+6 8.321 0.487
Cl syn convex AM1 LUMO+4 6.716 0.642
Br anti convex PM3 LUMO+6 8.991 0.548
Br syn convex PM3 LUMO+6 6.972 0.502
Br anti convex AM1 LUMO+6 8.323 0.624
Br syn convex AM1 LUMO+6 6.452 0.450
I anti convex PM3 LUMO+6 9.021 0.543
I syn convex PM3 LUMO+6 6.826 0.515
I anti convex AM1 LUMO+6 8.295 0.671
I syn convex AM1 LUMO+5 5.836 0.516
Graph 6
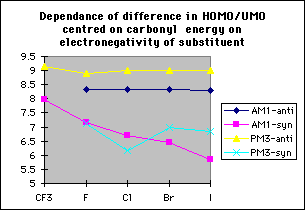
This can be rationalised using perturbation theory; the molecular orbitals of the two reactants were studied to see how they influence the transition state. This approach is applicable as the Hammond postulate states that for exothermic reactions the transition states are reactant-like. The calculated energy difference between the HOMO and the UMO centred on carbonyl for antiperiplanar attack is independent of the electronegativity of the substituent at the alpha-position; however for synperiplanar attack this energy difference decreases with decreasing electronegativity, increasing the perturbation stabilisation11 (table 2, graph 6). Therefore the difference in perturbation stabilisation, and hence the difference in transition state energies, for the two stereoisomers also decreases with decreasing electronegativity.
For attack from the convex face the calculated discrimination in favour of antiperiplanar attack is 4.46 or 2.13 kcal mol-1 at the PM3 or AM1 levels respectively. The calculated entropy difference between the two transition states of the two -fluoropenicillin stereoisomers of -6.78 cal K-1 mol-1 or 0.53 cal K-1 mol-1 at the PM3 or AM1 levels respectively results in a free energy difference of 2.43 kcal mol-1 or 2.29 kcal mol-1 at 300K. Energy differences of this magnitude correspond to a anti/syn ratio of ~60:1 and ~50:1 respectively.
For attack from the concave face the difference in energy between the transition states for the two isomers is lowered to 5.3 kcal/mol at the AM1 level. The discrimination and the entropy difference between the two stereoisomers cannot be calculated as a ground state could not be found for one of the isomers; hence the free energy difference and an anti/syn ratio could not be calculated. It would however be anticipated that these too would be lowered as the difference in energy of the transition states is lowered and hence the enthalpy difference is likely to be lowered; the difference in entropy is much smaller in magnitude than the difference in enthalpy for the two isomers, and hence the entropy difference has little effect on the calculated free energy difference or anti/syn ratio.
Although it would be expected that the calculated discrimination of the beta-lactamase would be much greater than that for lipases[5] as the calculated difference activation energies between the two stereoisomers isomers is much greater[12] ; it can be seen that the free energy difference and hence the anti/syn ratio is lower for the -lactamase than for the lipase[13]. This is because the entropy difference between the two isomers is much increased[14] indicating that the reactant ground states are much more organised.
Further modelling was carried out to establish whether the magnitude of the difference in transition state energies for the two stereoisomers is an artefact or a true result, which could indicate that another stabilising effect may be present.
Table 3: Results of modelling reaction of methoxide anion with cyclic and acyclic alpha-fluoroesters and lactams
Molecule Direction Hamiltonian TS O-C TS TS HOMO/ Coeff
of used bond Energy energies LUMO on
hydroxyl length /kcal of the Energy Carbonyl
attack /A two Difference Carbon
isomers
ester anti PM3 2.143 -208.660 2.345 9.350 0.185
ester syn PM3 2.369 -206.315 0.515
ester anti AM1 2.710 -220.335 -0.060 8.070 0.547
ester syn AM1 2.719 -220.395 8.043 0.547
amide anti PM3 2.368 -163.366 -3.037 8.407 0.564
amide syn PM3 2.186 -166.403 8.944 0.238
amide anti AM1 1.958 -170.335 -4.106 - -
amide syn AM1 2.008 -174.441 - -
beta-lactone anti PM3 2.220 -153.818 10.830 9.150 0.476
beta-lactone syn PM3 2.603 -142.988 7.408 0.576
beta-lactone anti AM1 2.500 -151.789 9.560 8.359 0.600
beta-lactone syn AM1 2.708 -142.230 7.606 0.600
beta-lactam anti PM3 2.212 -118.512 3.758 9.148 0.509
beta-lactam syn PM3 2.247 -114.755 8.907 0.548
beta-lactam anti AM1 2.354 -110.440 7.031 8.757 0.584
beta-lactam syn AM1 2.506 -103.409 8.097 0.576
penicillin anti PM3 2.291 -194.478 12.008 - -
penicillin syn PM3 2.763 -182.469 - -
penicillin anti AM1 2.581 -178.527 8.848 - -
penicillin syn AM1 2.671 -169.679 - -
In the model reaction of the methoxide anion with amides the discrimination between stereoisomers is decreased compared to that with esters, indicating that the presence of the nitrogen adjacent to the carbonyl instead of an oxygen results in a loss of discrimination (table 3). However the calculated discrimination is significantly increased in the model reactions of a lactone or lactam compared to acyclic esters and amides, showing that ring size might have a significant effect.
This can be rationalised using perturbation theory. Five main orbitals have to be considered when invoking the HOMO and LUMO: The unoccupied bonding orbitals on the carbonyl C and O, and on the methoxyl oxygen, the lone pair on the nitrogen and the unoccupied C-F n-[sigma]* antibonding orbital (scheme 3). The extent to which these orbitals combine to give the frontier molecular orbitals is determined by their orientation. Although other orbitals can combine if correctly orientated, if the molecular orbital is delocalised over too many atoms the contribution each atomic orbital is making cannot be determined. An alternative approach is to generate localised orbitals; these are an alternative solution to the wave function and are dispersed over a maximum of three atoms.
Scheme 3
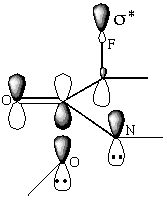
The occupied bond on the nitrogen has a destabilising effect on the frontier molecular orbitals; therefore the perturbation energy, and hence the discrimination between the stereoisomers, is lower for amides than for esters. As the orientation of the lone pairs on the oxygen is less rigid, these are unlikely to participate in the frontier molecular orbitals. The coefficient of the LUMO on the carbonyl carbon is increased for cyclic amides and esters compared to that for acyclics; hence the perturbation stabilisation is increased[11]. The particularly low LUMO coefficients of the carbonyl carbon, calculated using the PM3 hamiltonian for the antiperiplanar attack on the ester and the synperiplanar attack on the amide, arise because the favourable orientation of the carbon chain allows the unoccupied C-C n-[sigma]* antibonding orbitals to participate in the frontier molecular orbitals (diagram 1) The HOMO/LUMO energy difference appears to have little dependence on whether the ester or lactam is cyclic or not. The addition of an auxiliary ring also resulted in an increase in discrimination.
|  |
|---|---|
| LUMO of amide (syn attack) | LUMO of ester (anti attack) |
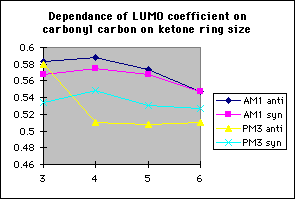
In order to study the effect ring size has on discrimination, the model reactions of the methoxide anion with 3,4,5 and 6 membered cyclic alpha-fluoroketones and alpha-fluorolactams were studied (tables 4 and 5 respectively)
Table 4: Results of modelling the reaction of methoxide anion with ketones of varying ring size
Ketone Direction Hamiltonian TS O-C TS Energy TS HOMO/ Orbital
Ring Size of used bond /kcal energy LUMO Coeff
hydroxyl length of the Energy on
attack /A two Difference Carbonyl
isomers Carbon
/kcal
3 anti PM3 2.555 -92.803 5.644 7.823 0.580
3 anti PM3 2.552 -87.160 7.404 0.535
3 syn AM1 2.575 -93.053 7.317 7.769 0.583
3 syn AM1 2.594 -85.736 7.467 0.568
4 anti PM3 2.361 -120.802 6.20331 8.297 0.511
4 anti PM3 2.462 -114.599 7.832 0.549
4 syn AM1 2.491 -119.734 7.67405 8.135 0.588
4 syn AM1 2.586 -112.060 7.649 0.575
5 anti PM3 2.335 -140.511 2.754 8.352 0.508
5 anti PM3 2.224 -137.757 8.897 0.531
5 syn AM1 2.445 -148.125 6.770 8.321 0.574
5 syn AM1 2.479 -141.355 7.951 0.567
6 anti PM3 2.218 -145.154 2.502 9.037 0.510
6 anti PM3 2.231 -142.652 8.946 0.527
6 syn AM1 2.629 -152.946 2.822 7.835 0.548
syn AM1 2.533 -150.124 7.876 0.548
6
Table 5: Results of modelling reaction of methoxide anion with lactams of varying ring size
Lactam Direction Hamiltonian TS O-C TS TS energy HOMO/ Orbital
ring size of used bond Energy of the two LUMO Coefficient
hydroxyl length /kcal isomers Energy on
attack /A /kcal Difference Carbonyl
Carbon
3 anti PM3 - - - - -
3 anti PM3 - - - -
3 syn AM1 2.485 -89.282 5.867 8.171 0.567
3 syn AM1 2.497 -83.416 7.931 0.579
4 anti PM3 2.212 -118.512 3.758 9.148 0.509
4 anti PM3 2.247 -114.755 8.907 0.548
4 syn AM1 2.354 -110.440 7.031 8.757 0.584
4 syn AM1 2.506 -103.409 8.097 0.576
5 anti PM3 2.307 -139.532 4.846 8.616 0.490
5 anti PM3 2.375 -134.686 8.205 0.529
5 syn AM1 2.373 -139.900 6.233 8.806 0.568
5 syn AM1 2.438 -133.667 8.305 0.543
6 anti PM3 - - - - -
6 anti PM3 2.782 -125.243 6.488 0.492
6 syn AM1 - - - - -
syn AM1 2.580 -127.900 7.513531 0.47495
6
Graph 7
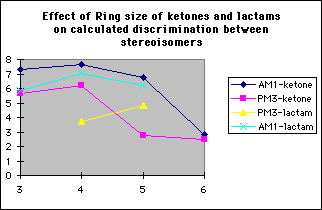
The model reactions of the methoxide anion with cyclic alpha-fluoroketones and alpha-fluorolactams indicate that discrimination between the stereoisomers is maximised for four membered rings, and decreases with increasing ring size (graph 7). The calculated discrimination of the methoxide anion for three membered rings is imbetween that of the four and five membered rings. This correlates to the magnitude of the LUMO coefficient (tables 4&5, graphs 8&9) on the carbonyl carbon and hence the perturbation stabilisation11. The HOMO/LUMO energy difference appears to have little dependence on ring size. The calculated discriminations for the 4 and 5 membered ring lactams using the PM3 hamiltonian are anomalous; the unoccupied C-F n-[sigma]* antibonding orbital is orientated so that it can participate in the LUMO for synperiplanar attack, although to not the same extent as for antiperiplanar attack, hence reducing the difference in perturbation stabilisation between the two isomers (diagram 2).
| 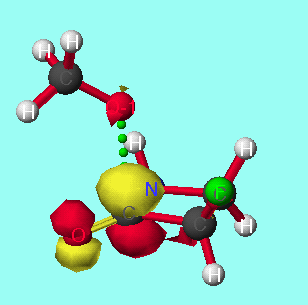 |
|---|---|
| `Antiperiplanar' methoxyl attack | `Synperiplanar' methoxyl attack |
Graph 8
Graph 9
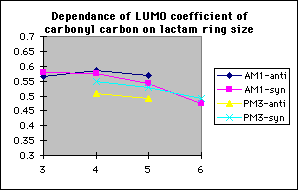
Table 6: Results of modelling reaction of methoxide anion with thioketones of varying ring sizes
Thioketone Direction Hamiltonian TS O-C TS TS energy HOMO/ Orbital
Ring Size of used bond Energy of the two LUMO Coeff
hydroxyl length /kcal isomers Energy on
attack /A /kcal Difference C-S
Carbon
3 anti PM3 2.272 -24.998 -1.455 6.314759 0.56074
3 anti PM3 2.293 -26.370 6.270127 0.56660
3 syn AM1 2.334 -31.884 -1.238 7.831298 0.42236
3 syn AM1 2.342 -33.122 6.584232 0.46335
4 anti PM3 2.344 -48.351 - 6.864673 0.58037
4 anti PM3 - - - -
4 syn AM1 2.624 -53.908 17.093 6.456139 0.57291
4 syn AM1 2.452 -36.815 7.610535 0.37548
5 anti PM3 2.280 -67.391 4.220 6.375095 0.54764
2.527 -68.714
5 anti PM3 2.364 -63.171 6.556471 0.56862
5 syn AM1 2.556 -82.063 6.121 6.902105 0.57550
5 syn AM1 2.598 -75.942 6.658734 0.54508
6 anti PM3 2.281 72.093 6.994 7.526655 0.57723
6 anti PM3 2.348 -65.099 6.61921 0.56356
(2.119) -70.661
6 syn AM1 2.260 -87.810 6.200 8.014387 0.55509
syn AM1 2.602 -81.610 6.60995 0.54874
6
Graph 10
Graph 11
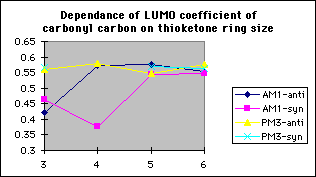
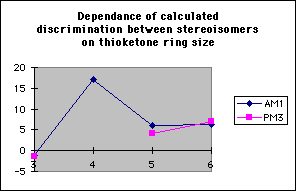
If the carbonyl is replaced by a thioketone, modelling indicates that ring size has a similar effect on the calculated discrimination between the stereoisomers, with discrimination being maximised for four membered rings, although the actual magnitude of this discrimination appears to be less predictable (table 6, graph 10).The dependence of the LUMO coefficient on the carbonyl carbon on ring size is less predictable (graph 11), and hence the perturbation stabilisation is less dependant on ring size. The 3sp2 orbitals of the sulfur overlap less well with the sp3 carbon orbitals than the 2sp2 orbitals of oxygen, hence the C-S bond has less double bond character and the extent of the participation of the C-S bonding orbital in the frontier molecular orbitals is less predictable. The coefficient on the C-S carbon for synperiplanar attack on a four membered ring thioketone, calculated using the AM1 hamiltonian is anomalous as an O-H [sigma]* antibonding orbital is orientated so that it can participate in the LUMO, reducing the coefficient on the C-S carbon and increasing the perturbation stabilisation11 (diagram 3)
Diagram 3: LUMO corresponding to synperiplanar attack calculated using the AM1 hamiltonian
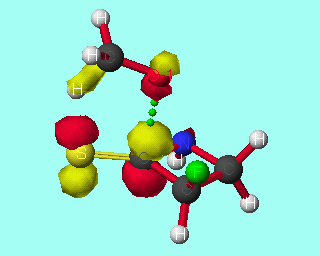
Scheme 4
Table 7: Results of modelling reaction of reaction of methoxide anion with ketones with nitrogen substituted at varying ring positions
Ketone Posn Direction Hamiltonian TS O-C TS TS energy HOMO/ Orbital
Ring of N of used bond Energy of the LUMO Coeff
Size in Ring hydroxyl length /kcal two Energy on
attack /A isomers Difference Carbonyl
/kcal /eV Carbon /eV
4 2 anti PM3 2.297 -109.558 5.588 8.494 0.519
4 2 syn PM3 2.312 -103.971 8.347 0.560
4 2 anti AM1 2.833 -95.851 8.685 7.339 0.597
4 2 syn AM1 2.605 -87.166 7.422 0.579
5 2 anti PM3 2.352 -132.456 5.360 8.252 0.530
5 2 syn PM3 2.398 -127.096 7.910 0.552
5 2 anti AM1 2.366 -134.584 7.074 8.547 0.583
5 2 syn AM1 2.484 -127.510 7.928 0.573
5 3 anti PM3 2.355 -135.835 5.096 8.265 0.547
5 3 syn PM3 2.418 -130.739 7.875 0.562
5 3 anti AM1 2.399 -132.274 4.274 8.447 0.590
5 3 syn AM1 2.453 -128.000 8.082 0.578
6 2 anti PM3 - - - - -
6 2 syn PM3 - - - - -
6 2 anti AM1 - - - - -
6 2 syn AM1 2.565 -128.141 8.309 0.585
6 3 anti PM3 - - - - -
6 3 syn PM3 - - - - -
6 3 anti AM1 - - - - -
6 3 syn AM1 2.620 -126.181 7.415 0.563
6 4 anti PM3 2.334 -135.072 7.131 8.271 0.420
6 4 syn PM3 2.428 -127.941 - 7.567 0.516
6 4 anti AM1 2.438 -136.247 10.218 8.170 0.552
6 4 syn AM1 2.589 -126.029 - - -
Table 8: TS energy of the two isomers for ketones with N substituted at varying ring positions /kcal
Ring Size
Position of 3 4 5 6
N in ring PM3 AM1 PM3 AM1 PM3 AM1 PM3 AM1
1 5.867 3.758 7.031 4.846 6.233 - -
-
2 - - 5.588 8.685 5.360 7.074 - -
3 - - - - 5.096 4.274 - -
4 - - - - - - 7.131 10.218
Table 9: LUMO Coefficient on Carbonyl Carbon for ketones with N substituted at varying ring positions /eV
Ring Size
Position of 3 4 5 6
N in ring PM3 AM1 PM3 AM1 PM3 AM1 PM3 AM1
For antiperiplanar hydroxyl attack
1 - 0.567 0.509 0.584 0.49 0.568 - -
2 - - 0.519 0.597 0.530 0.583 - -
3 - - - - 0.547 0.590 - -
4 - - - - - - 0.420 0.552
For synperiplanar hydroxyl attack
1 - 0.579 0.548 0.576 0.529 0.544 0.492 0.475
2 - - 0.560 0.579 0.552 0.573 - 0.585
3 - - - - 0.562 0.578 - 0.563
4 - - - - - - 0.516 -
Table 10: HOMO/LUMO Energy Difference for ketones with N substituted at varying ring positions /eV
Ring Size
Position of 3 4 5 6
N in ring PM3 AM1 PM3 AM1 PM3 AM1 PM3 AM1
For antiperiplanar hydroxyl attack
1 - 8.171 9.148 8.757 8.616 8.806 - -
2 - - 8.494 7.339 8.252 8.547 - -
3 - - - - 8.265 8.447 - -
4 - - - - - - 8.271 8.170
For synperiplanar hydroxyl attack
1 - 7.931 8.907 8.097 8.205 8.305 6.488 7.514
2 - - 8.347 7.422 7.910 7.928 - 8.309
3 - - - - 7.875 8.082 - 7.415
4 - - - - - - 7.567 -
The discrimination between stereoisomers is maximised if the nitrogen is beta- to the carbonyl in the ring, and decreases with increasing distance of the nitrogen from the carbonyl. If the nitrogen is alpha- to the carbonyl the discrimination between the stereoisomers is also less than that for beta-substituted rings (tables 7&8). The LUMO coefficient on the carbonyl carbon increases (table 9), while the HOMO/LUMO energy difference decreases (table 10), with increasing distance of the nitrogen from the carbonyl. Hence the perturbation stabilisation should increase with increasing distance of the nitrogen from the carbonyl. If the nitrogen is alpha- to the carbonyl, the occupied bond on the nitrogen has a destabilising effect on the frontier molecular orbitals; therefore the perturbation energy is increased and the discrimination between the stereoisomers is lowered. However, if the nitrogen is beta- to the carbonyl in the ring, the unoccupied C-N n-[sigma]* antibonding orbital can also combine to give the frontier molecular orbitals, increasing the perturbation stabilisation (scheme 4). If the distance between the nitrogen and the carbonyl is increased further, the nitrogen atomic orbitals are not sufficiently close to the bond forming to make any significant contribution to the molecular orbitals, and the perturbation stabilisation is lowered.
Scheme 5
| 
| 
|  Reactant ground state
| Hypobromite
| Tetrahedral intermediate
| Product ground state | |
|---|
Table 11: Results of modelling the reactant, intermediate and product ground states for reaction of methoxide anion with alpha-halopenicillins
Group Direction Hamilton Reactant Reactant O-C Tetrahedral Product Product
substitu of hydroxyl ian Used O-C bond bond Intermediate O-C Energy
ted at group attack length /A Energy length Energy/kcal Bond /kcal
alpha-pos /kcal intermed length/
/A A
CF3 anti convex PM3 3.684 -322.73 1.429 -344.811 3.392 -343.873
CF3 syn convex PM3 5.408 -294.39 1.422 -351.342 2.765 -346.533
CF3 anti convex AM1 3.567 -294.29 1.437 -328.478 3.502 -345.200
CF3 syn convex AM1 6.497 -292.73 1.429 -332.968 2.762 -346.729
F anti convex PM3 3.699 -206.19 1.428 -230.236 3.522 -227.752
F syn convex PM3 4.390 -198.65 1.420 -233.947 2.708 -232.777
F anti convex AM1 3.823 -183.56 1.438 -217.112 3.465 -232.632
F syn convex AM1 4.585 -176.85 1.429 -220.173 2.779 -234.813
F anti concav PM3 5.275 -200.24 ring - 2.697 -230.194
opened
F syn concav PM3 3.060 -210.13 1.419 -228.474 2.868 -232.243
F anti concav AM1 3.880 -182.60 1.443 -214.491 3.315 -232.566
F syn concav AM1 12.092 -168.54 1.452 -216.422 2.979 -231.469
Cl anti convex PM3 3.728 -168.87 1.429 -191.583 2.893 -192.161
Cl syn convex PM3 4.361 -160.51 1.420 -194.547 2.816 -196.067
Cl anti convex AM1 3.751 -144.17 1.440 -177.479 2.816 -192.074
Cl syn convex AM1 formed - 1.428 -178.939 2.816 -195.759
hypochlorite
Br anti convex PM3 3.712 -156.64 1.427 -179.453 2.798 -182.060
Br syn convex PM3 formed - 1.416 -184.490 2.818 -187.642
hypobromite
Br anti convex AM1 3.790 -132.16 1.439 -164.966 2.787 -179.829
Br syn convex AM1 formed - 1.427 -165.636 2.962 -182.546
hypobromite
I anti convex PM3 3.576 -133.54 1.427 -155.064 2.833 -160.345
I syn convex PM3 5.525 -128.26 1.413 -163.815 2.694 -166.527
I anti convex AM1 3.784 -120.01 1.439 -153.028 2.775 -168.807
I syn convex AM1 formed - - - - -
hypoiodite
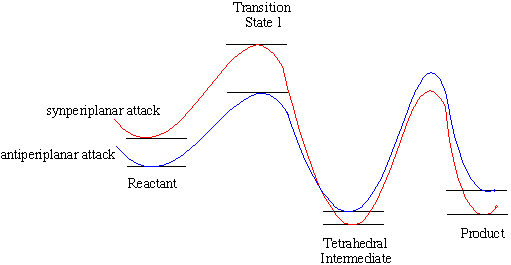
For attack from both the convex and the concave face, if the reaction is carried out under kinetic control, antiperiplanar attack is favoured as the transition state energy is lower. If the reaction is carried out under thermodynamic control, synperiplanar attack is favoured, as the tetrahedral intermediate and product energies are lower (scheme 3).
This is also normally seen with other halogen substituted penicillins and the penicillin substituted with the CF3 moiety at the alpha position.
The methoxide anion (serine analogue) is far more basic than the serine 70 hydroxide of a beta-lactamase and hence sometimes acted as a base instead of a nucleophile in simulations. Hypohalides were also sometimes preferentially formed when attempting to simulate the reactant ground states for synperiplanar attack of the methoxide on penicillins alpha-substituted with less electronegative substituents.
The presence of hydrogen bonding in the reactant ground state of the antiperiplanar isomers could account for the large entropy difference seen between the two stereoisomers[30], as the reactant ground state corresponding to antiperiplanar attack is particularly ordered.
Table 12: Results of QUEST search of the Cambridge crystallographic database[15] for organics only with a non-bonded contact between a hydrogen alpha- to a carbonyl and an ether, hydroxyl or carbonyl oxygen between 1.5 and 2.0A
Molecule Reference Reasons for H Type of Length
acidity in O of O-H
addition to involved Hydrogen
carbonyl in H Bond /A
bonding
Alanyl-proline-4-nitroanilide 17 - to N carbonyl 1.977
hydrochloride monohydrate
(2aR,4S,4aS,5S,7aS,7bR)-Octahydro-2H, 18 - carbonyl 1.677
5H-1,4,7-trioxacyclopent(j,k,l)-as-in
dacen-5-one
1,3-Selenazolidine-2,4-dione 19 on lactam lactam 1.712
DL-threo-beta-Fluoroaspartic acid 20 alpha to F carbonyl 2.037
dihydrate
5,8-Dihydroxy-1,4-naphthoquinone 21 - carbonyl 1.935
(Naphthazarin form A) 2.022
Deoxycholic acid acetone at 103 deg.K 22 - carbonyl 1.576
1.616
1.889
5-(5-(2',6'-Di-iodo-4'-methylphenoxy) 23 on lactam lactam 1.782
benz-2-olyl)-2-pyridone
6-Amino-1,6-dideoxy-1-(3,4-dihydro-3- 24 on ester ester 1.970
methyl-2,4-dioxo-1(2H)-pyrimidinyl)
4-thio-L-glycero-alpha-L-ido-heptofur
anuronic acid monohydrate
9balpha-Methyl-2,3,3aalpha,4,5,5abeta 25 H on tertiary carbonyl 1.882
,6,7,8,9,9aalpha,9b-dodecahydro-1H-cy C
clopenta(a)naphthalene-1,5-dione
Pentaerythritol tetra-acetate 26 on ester ester 1.546
Expansolide A expansolide B 27 - carbonyl 1.986
(3S,8aS)-3-Bromomethyl-3-methyl-1,4-d 28 - to N ester 1.937
ioxo-3,4,6,7,8,8a-hexahydro-1H-pyrrol
o(2,1-c)(1,4)oxazine
6-Hydroxyflavone 29 - to ether O carbonyl 1.805
The X-ray crystallographic structure of a beta-lactamase co-crystallised with a phosphonate inhibitor was obtained from the Brookhaven protein data bank.
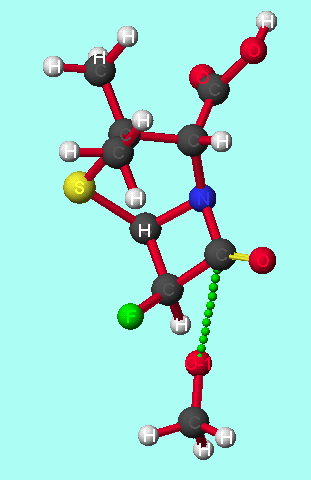
Diagram 4

-LACTAMASE (E.C.3.5.2.6) COMPLEXED WITH [[N-(BENZYLOXYCARBONYL)AMINO]METHYL]PHOSPHONATE[31]
The phosphonate inhibitor is an analogue of the transition state/ intermediate of beta-lactam hydrolysis; hence it should be possible to directly replace the inhibitor with the calculated transition states and calculate the energies of each complex, using the program QUANTA[32]. Comparison of these energies should confirm the results obtained through modelling on which is the most favoured direction of attack; whether it is anti- or syn-periplanar and is from the convex or concave face of the penicillin.
The modelling studies undertaken so far during this project have considered only the stereoelectronic effects at the active site; electrostatic and steric effects have been ignored. Any steric effects at the active site caused by replacing the hydrogen with a halogen need to be considered, in addition to any hydrogen bonding set up between the residues at the active site and the ester on the five membered ring of the transition state. CHARMm[33] energy calculations performed on the transition states docked into the enzyme should indicate the preferred direction of attack by the Ser70 residue. CHARMm[33] energy minimisations should allow the tertiary structure of the -lactamase at the active site to relax and form any favourable electrostatic interactions, and minimise unfavourable steric interactions. Analysis of these structures would indicate whether the halogen has a significant steric effect and whether any novel hydrogen bond interactions are set up.
Diagram 5
Transition state corresponding to antiperiplanar attack from the convex face by the ser70 residue, calculated using the PM3 hamiltonian, docked into the active site of the beta-lactamase.
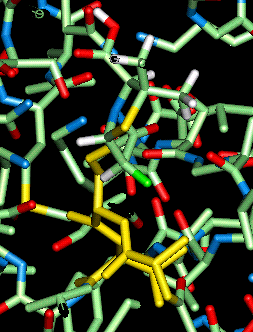
However, a lot of difficulty was experienced in calculating the minimum energies of the enzyme with a calculated transition state docked instead of the phosphonate inhibitor. Despite many communications with MSI support and a QUANTA expert from GlaxoWellcome no value for the minimum energy could be calculated; the QUANTA[32] communication with the energy calculation program CHARMm[33] always became inactive.
It was also attempted to use the program MACROMODEL[34] to calculate the minimum energies for the transition state docked into the beta-lactamase. However, as a very large number of parameters were missing from the forcefield, that would have had to be estimated or calculated, this alternative approach was abandoned.
David O'Hagan, at the university of Durham, is currently undertaking an experimental study into the stereoselectivity of beta-lactamases in reactions with alpha-fluorosubstituted penicillins. When these results are available, the simulation and experimental results can be compared.