| [Related articles/posters: 028 011 005 ] |
Deirde M.A. Conlon and David A. Widdowson*
Department of Chemistry, Imperial College of Science, Technology and Medicine, London SW7 2AY, UK.Abstract
A synthetic approach to the ergoline system based on, inter alia , chromium mediated 4-functionalisation of indole and palladium catalysd couplings of an intact ring D moiety is described. The sequence has been developed to the advanced 4,5-seco -16-norergoline stage.
Introduction
The synthesis of 4-substituted indoles is a major target of natural product and pharmaceutical compound synthesis1. Because of the difficulties of direct 4- substitution in indoles2 most approaches involve the synthesis of the indole system presubstituted at C-4 by construction of either the pyrrolic ring (Scheme 1a)3 or the benzenoid ring (Scheme 1b)4 in a manner which incorporates an appropriate function, R, at C-4.
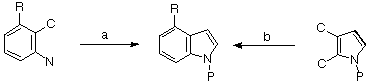
Scheme 1
Direct functionalisation at C-4 on the intact indole ring has been achieved, in a restricted manner, by intramolecular cyclisation from C-3, in a suitably activated and C-2 blocked substrate (Scheme 2a)5, or, in a more general process, by directed metallation, notably by thallium, using a C-3 function at the directing group (Scheme 2b)6. In each case the substituent at C-3 is essential to the process.
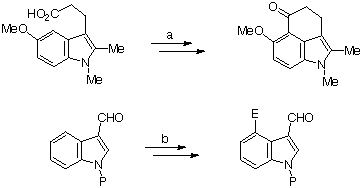
Scheme 2
The direct synthesis of 4-substituted indoles without a 3-substituent has been achieved by activation of the benzenoid ring to metallation of addition by attachment of a transition metal to that ring7. Of the metals used, the tricarbonylchromium(0) moiety has received most attention and an extensive chemistry of indoletricarbonylchromium(0) complexes have been reported8. The electron deficiency induced in the ring by the metal carbonyl unit renders it susceptible to both nucleophilic addition (Scheme 3a)9 and to deprotonation (Scheme 3b)10 and both of these approaches have been used to produce 4-substitution11.
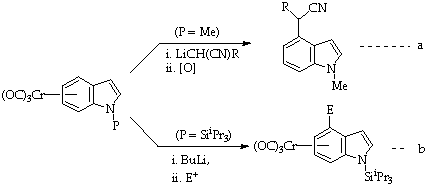
Scheme 3
We have sought to exploit the approaches generalised in Schemes 1a and 3b, in order to gain rapid access to 4-substituted indoles which contain a preformed ringD of the ergoline molecular framework and so develop the short synthesis of the ergoline system. We had established that palladium catalysed coupling to C-4 of indoles could be seriously inhibited by functionality at C-312, presumably by a sterically constraining peri-interaction. It was planned, therefore, to introduce the indole 4-substituent prior to 3-functionalisation and this dictated that carbon 4 (of the ergoline framework) must be introduced at a late stage, either by attachment at C-3 followed by closure on to ring D or by incorporation of this carbon in the ring D fragment. For our initial approach, we chose the former as expressed retrosynthetically in Scheme 4.
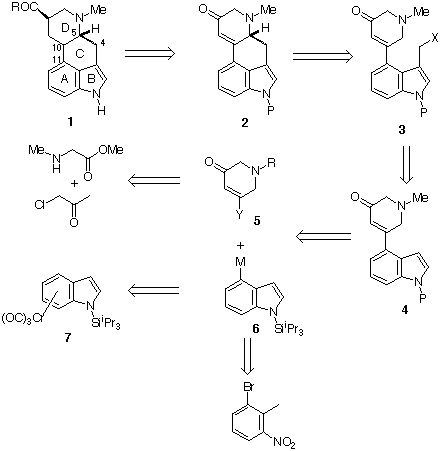
Scheme 4
We descibe here the synthesis of the advanced 4,5-seco -16-norergoline intermediate 3, X = NMe2, P = H, of Scheme 413.
Discussion
4-Substituted Indole Synthesis.
4-Bromoindole was best prepared by a modification of the Leimgruber-Batcho synthesis14. Thus 2-bromo-6-nitrotoluene was heated in toluene with DMF dimethyl acetal, DMF and pyrrolidine at 110°C for 2 h. The resultant enamine was reduced with titanium trichloride - ammonium acetate in methanol15 to give 4-bromoindole in 71%yield. Silylation of this with TIPS chloride - sodium hydride gave the protected indole 6, M = Br, (81%) required for the ring D coupling step. Lithiation of this (BuLi, THF) and quenching with triisopropyl borate or trimethylchlorostannane gave after work up, respectively, the analogous boronic acid 6, M = B(OH)2, (80%) or the stannane 6, M = SnMe3 (83%) (Scheme 5a).
Alterrnatively, eta6-(1-triisopropylsilylindole)tricarbonylchromium(0) 7 was lithiated (BuLi, THF, &endash;78°C) and transmetallated with copper bromide-dimethylsulfide complex to generate the cuprate 8, M = CuLn, P = SiiPr3 (Scheme 5b)16 which was used without isolation in coupling processes.
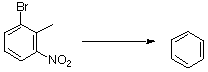
Scheme 5
Assembly of the ring D fragments.
The basic ring D fragment 5, Y = OH was synthesised in 35% overall yield from sarcosine according to Shepherd17 in both the N-methyl series (R = Me) and the N-benzyl series (R = Bn). These were converted to the analogous sulfonates or halides (Cl, Br) as shown in Scheme 6.
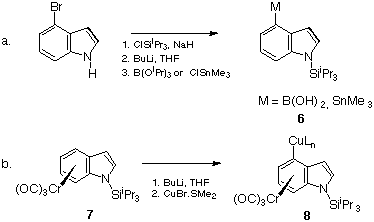
Scheme 6
Thus the mesylate 5, Y = OSO2Me (89%), the tosylate 5, Y = OSO2C6H4Me (42%), and the triflate 5, Y = OSO2CF3 (31%) were prepared as unstable yellow oils, which darkened rapidly at room temperature. The halides of the N-benzyl series were best made by displacement of sulfonate under Lewis acidic conditions (BF3.OEt2, BnNMe3+ Hal&endash;)18 which generated the chloride 5, R = Bn, Y = Cl (46%) and bromide 5, R = Bn, Y = Br (31%) again as unstable yellow oils. The analogous N-methyl compounds were only isolable in 10% yield and not further used.
Coupling of ring D to ring A.
With a variety of the ring A and ring D fragments available, the coupling of these by a tetrakistriphenylphosphinepalladium catalysed process19 was examined. A test reaction with 3-chlorocyclohex-2-enone and complex 8, followed by decomplexation of the product with air, gave a total conversion of 74% to the product 9 (60%) together with the desilylated analogue 10 (14%) (Scheme 7a).
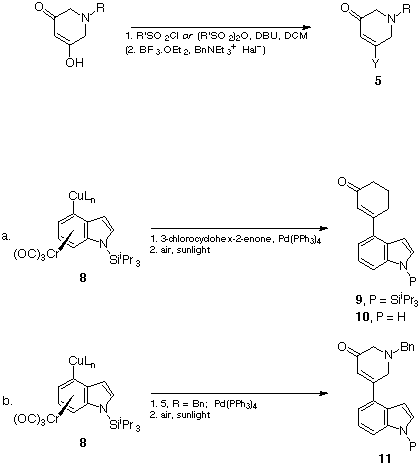
Scheme 7
The analogous reactions with the tetrahydropyridone series 5, R = Bn gave the coupled, desilylated product 11, P = H in 45%yield, together with some N-silylated material 11, P = SiiPr3 (5%) and 4-butylated byproduct (5%).
The equivalent reaction of the cuprate derived from the uncomplexed indoles 6, M = Br, with the N-benzyltetrahydropyridones 5, gave the same indolic product 11, P = SiiPr3 (45%) in a cleaner process. Desilylation (TBAF, THF) then gave the indole 11, P = H (99%). This sequence therefore became the preferred route to the compounds 11 and 4.
With the ring A &endash; ring D coupling process established, the required N-methyl series was introduced to the sequence. Coupling of the 4-metallated N-TIPS indoles 6, M = CuLn or B(OH)2 or SnMe3, generated previously, with the pyridones 5, R = Me gave the product 4, P = SiiPr3 in 31%, 60% and 30% respectively (Scheme 8). The Suzuki type coupling20 of the boronic acid was therefore established as the preferred method for subsequent application.
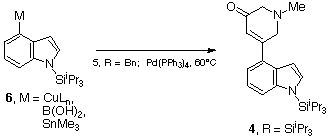
Scheme 8
Synthesis of 4,5-seco -16-norergoline.
The introduction of the one carbon fragment at C-3 was envisaged as an electrophilic coupling of 4 with Eschenmoser's reagent (Me2N+=CH2 I&endash;)21. Initially, a series of simple 4-substituted indoles 6, M = H, CH2OAc, CHO, CH=CHOMe, were examined and found to couple at C-3 with high efficiency (98%, 98%, 91% and 100% respectively) (Scheme 9a).
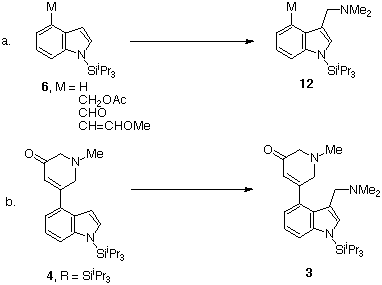
Scheme 9
The N-benzyl series analogue 11, however, failed to react, either with or without the 1-SiiPr3 group. In the methyl series, no reaction occurred if the N-silyl group was present but in situ aminomethylenation of the (presumed) anionic product of fluoride ion desilylation gave the gramine analogue 3, X = NMe2, P = H in 22% yield (Scheme 9b). A detailed study of this reaction has failed to raise the yield to a more acceptable level. Thus far, all attempts to generate the C4&endash;C5 bond (ergoline numbering) have failed and alternative strategies are under development.
By this means the advanced intermediates 4 and 3 have been prepared in 3 and 4 steps respectively from the readily available 4-bromoindole in 39% and 9% respectively.
Experimental
4-Bromoindole.14,15
A mixture of 2-bromo-6-nitrotoluene (100 g, 460 mmol), dimethylformamide-dimethyl acetal (184 ml, 1.38 mmol) and pyrrolidine (39 ml, 460 mmol) in DMF (1 l), was heated at 110°C for 3 h. The cooled mixture was poured into a 1 : 1 mixture of ice and water (1.5 l) and extracted with diisopropyl ether (3 x 1000 ml) until little red colour remained in the aqueous layer. Solvent removal in vacuo gave the intermediate 1-(N,N -dimethyl)-2-(2-bromo-6-nitrophenyl)ethenylamine (120 g). This (5.4 g, 20 mmol) , in methanol, (50 ml), was added dropwise to a mixture of titanium trichloride (10% solution in 20-30% hydrochloric acid, 120 ml, 77 mmol) and aqueous ammonium acetate (4 M. 240 ml, 960 mmol) in a 1 l. dropping funnel and the mixture shaken vigorously for 10 min. Dilute hydrochloric acid (10%, 25 ml) was added to the mixture which was then extracted with diisopropyl ether (2 x 200 ml). The combined organic extracts were washed with water (250 ml), dried (MgSO4) and concentrated in vacuo to give a brown oil which was purified by column chromatography (SiO2, 3:1 hexane:dichloromethane). 4-Bromoindole was obtained as a yellow oil, (2.9 g, 71%) b.p. 91-93°C/0.5 mmHg (lit.22 96°C/1.0 mmHg), i.r. (film)/cm-1 3415, 1620, 1615, 1570, 1430, 1340, 1175, 890, 755; p.m.r. (CDCl3) 6.59 (1H, m), 7.01 (2H, m), 7.09 (1H, m), 7.19 (1H, m), 8.18 (1H, br s, ).
1-Triisopropylsilylindoletricarbonylchromium(0) (6, M = H).
1-Triisopropylsilylindole (3.23 g, 12 mmol) and chromium hexacarbonyl (3.91 g, 18 mmol) were heated under reflux in a mixture of THF (10 ml) and dibutyl ether (120 ml) for 24 h., then cooled to rom temperature and filtered through Celite and the solvent was removed in vacuo . Addition of petroleum ether (b.p. 40-60°C, 50 ml) precipitated 1-triisopropylsilylindoletricarbonylchromium(0) (6, M = H) as a yellow crystalline solid (19.81 g, 42%), m.p. 135-136°C (lit.11 137-139°C); i.r. (Nujol)/cm-1 1950, 1870, 1410, 1035, 880; p.m.r. (CDCl3) 1.25 (18H, 2 x d, J 7.5Hz), 1.61 (3H, sept, J 7.5Hz), 5.10 (1H, t, J 7Hz), 5.31 (1H, t, J 7Hz), 6.20 (2H, br d, J 7Hz), 6.42 (1H, d, J 1Hz), 7.30 (1H, d, J 1Hz); m/z (EI) 409 (M+) 381, 353, 325, 273, 230 (100%).
1-Triisopropylsilylindole-4-boronic acid [6, M = B(OH)2].
Butyllithium (1M in hexanes, 2 ml, 2 mmol0 was added dropwise to a solution of 4-bromo-1-triisopropylsilylindole (0.66 g, 1.88 mmol) in THF (10 ml) at &endash;78°C. After 2 h, triisopropyl borate (1.2 ml, 5 mmol) was added, causing a significant exotherm. The mixture was allowed to warm to ambient temperature, quenched with saturated aqueous ammonium chloride (25 ml), extracted with ether and dried (MgSO4) and evaporated to drynessin vacuo . The crude residual oil was chromatographed (SiO2, ether:hexane 0:100 &endash;> 50:50) to give a mixture of 1-triisopropylsilylindole-4-boronic acid (6, M = B(OH)2) and its cyclic trimer (0.45 g, 80%) as a white foam, identified by their spectra: (6, M = B(OH)2) p.m.r. (400 MHz, CDCl3) 1.13 (18H, d, J 7.5Hz), 1.73 (3H, d, J 7.5Hz), 4.74 (2H, s), 6.99 (1H, dd, J 1, 3Hz), 7.18 (1H, t, J 7Hz), 7.36 (1H, d, J 3Hz), 7.45 (1H, dd, J 7, 1Hz), 7.66 (1H, br d, J 8Hz); trimer: p.m.r. (400 MHz, CDCl3) 1.19 (18H, d, J 7.5Hz), 1.75 (3H, d, J 7.4Hz), 7.34 (1H, t, J 7Hz), 7.49 (1H, br d, J 3Hz), 7.67 (1H, d, J 3Hz), 7.77 (1H, br d, J 8Hz), 8.25 (1H, br d, J 7Hz); m/z (EI) 898 (M+), 855. Found: M+&endash;iPr, 896.5547. C51H77N3O3Si3 requires 896.5552.
1-Triisopropylsilyl-4-trimethylstannylindole (6, M = SnMe3).
Butyllithium (1.3 M, 30 ml, 40 mmol) was added slowly to a solution of 1-triisopropylsilyl-4-bromoindole (14.1 g, 40 mmol) in THF (50 ml) at &endash;78°C. After 30 min. a solution of trimethyltin chloride (40 g, 200 mmol) in THF (20 ml) was added rapidly and the mixture stirred at &endash;78°C for a further 1 h. then warmed to room temperature and quenched with water (100 ml). The mixture was extracted with ether ( 2 x 100 ml) and the combined ethereal extracts washed with water and brine (200 ml of each), dried (MgSO4)and evaporated to dryness. The crude oil was purified by filtration through a short plug of alumina, with hexane as eluent, to give 1-triisopropylsilyl-4-trimethylstannylindole (6, M = SnMe3) (14.4 g, 83%) as colourless crystals, m.p. 38-40°C, i.r. (Nujol)/cm-1 1520, 1465, 1405, 1280, 1155, 885, 755; p.m.r. (CDCl3) 0.33 (9H, s), 1.14 (18H, d, J 7.5Hz), 1.70 (3H, sept, J 7.5Hz), 6.59 (1H, dd, J 3, 2Hz), 7.08 (1H, dd, J 8, 7Hz), 7.22 (2H, m), 7.49 (1H, br d, J 8Hz); m/z (EI) 437 (M+), 422, 392, 273, 59 (100%).
1-Benzyl-5-hydroxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = OH).
Chloroacetone (73 ml, 910 mmol) was added slowly to a mixture of N-benzylglycine ethyl ester (176 g, 910 mmol) and sodium hydrogen carbonate (77 g, 910 mmol) in acetonitrile (600 ml) and water (250 ml) at 70°C. Stirring was continued at this temperature for a further 6 h. then the solvents were removed in vacuo and the residue was partitioned between ether (500 ml) and water (250 ml). The aqueous phase was extracted with ether (200 ml0 and the combined ether phases were dried (MgSO4) and evaporated to dryness. The residue was distilled to give N-benzyl- N-(2-oxopropyl)glycine ethyl ester (148.4 g, 66%), b.p. 116-118°C / 0.1 mm Hg (lit.17b.p. 116-120°C / 0.1 mm Hg); i.r. (film)/cm-1 1725, 1450, 1190, 1150, 1025, 740, 700; p.m.r. (CDCl3) 1.26 (3H, t, J 7.1Hz), 2.11 (3H, s), 3.45 (2H, s), 3.52 (2H, s), 3.84 (2H, s), 4.42 (2H, q, J 7.1Hz), 7.26-7.43 (5H, m); m/z (EI) 249 (M+), 206,176, 91 (100%). Calc. for C14H19NO3: C, 66.45; H, 7.68; N, 5.62. Found: C, 66.85; H, 7.74; N, 5.70%.
A solution of sodium (3.6 g, 156 mmol) in ethanol (50 ml) was diluted with ether (100 ml) and N-benzyl- N-(2-oxopropyl)glycine ethyl ester (39 g, 156 mmol) added dropwise. The mixture was stirred at room temperature for 2 h., then quenched by dropwise addition of trifluoroacetic acid (12.5 ml, 160 mmol). The solvents were removed in vacuo and the residue dissolved in water and allowed to crystallise overnight to give 1-benzyl-5-hydroxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = OH) as a yellow powder (17.8 g, 56%), m.p. 79-80°C; i.r. (Nujol)/cm-1 3390(br), 1676, 1550, 1460, 1205, 830, 700; p.m.r. (200 MHz, d6 -DMSO) 3.1 (4H, s), 3.72 (2H, s), 5.31 (1H, s), 7.30 - 7.40 (5H, m).
1-Benzyl-5-methanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = OSO2Me).
Methanesulfonyl chloride (2 ml, 25 mmol) was added slowly to a suspension of 1-benzyl-5-hydroxy[1,2,3,6]tetrahydropyrid-3-one (4.88 g, 24 mmol0 and potassium carbonate (9.6 g, 70 mmol) in dichloromethane (50 ml) at room temperature. The mixture was stirred for 2 h., during which time the the it darkened. The mixture was diluted with dichloromethane (50 ml), then washed with water (2 x 50 ml) and and brine (50 ml). The organic phase was dried (MgSO4), filtered and concentrated in vacuo to give 1-benzyl-5-methanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = OSO2Me) as an unstable white amorphous powder (2.275 g, 38%); i.r. (Nujol)/cm-1 3100, 3040, 1665, 1620, 1450, 1360, 1200, 1120, 795, 730; p.m.r. (CDCl3) 3.20 (3H, s), 3.23 (2H, br s), 3.41 (2H, t, J 1Hz), 3.71 (2H, s), 6.15 (1H, t, J 1.3Hz), 7.31-7.35 (5H,m); m/z (EI) 281 (M+), 202, 120, 91 (100%). [Found: C, 55.0; H, 5.37; N, 4.88. C13H15NO4. 0.1H2O requires C, 55.15, H, 5.41; N, 4.95%].
1-Benzyl-5-bromo[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = Br).
Benzyltrimethylammonium bromide (0.359 g, 0.8 mmol) was added in one portion to a stirred solution of 1-benzyl-5-methanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (0.526 g, 0.8 mmol) in dichloromethane (10 ml) at room temperature. Boron trifluoride etherate (0.3 ml, 2.4 mmol) was added slowly and stirring continued for 1 h. The mixture was diluted with dichloromethane (25 ml) and poured into water (25 ml). The organic layer was washed with brine (25 ml), dried (MgSO4) and evaporated to dryness giving 1-benzyl-5-bromo[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = Br) (0.07 g, 13%) as a brown, unstable oil, p.m.r. (CDCl3) 3.25 (2H, br s), 3.59 (2H, br s), 3.71 (2H, s), 6.54 (1H, t, J 1Hz), 7.30-7.47 (5H, m);m/z (EI) 265/267 (M+), 186, 176/178, 91 (100%).
1-Benzyl-5-chloro[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = Cl).
Benzyltriethylammonium chloride (1.13 g, 5 mmol) was added in one portion to a stirred solution of 1-benzyl-5-methanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (1.235 g, 4.65 mmol) in dichloromethane (25 ml) at room temperature. Boron trifluoride etherate (1.5 ml, 12 mmol) was added slowly over 10 min. and stirring continued for 1 h. The mixture was diluted with dichloromethane (50 ml) and poured into water (50 ml). The organic layer was washed with brine (50 ml), dried (MgSO4) and evaporated to dryness to give 1-benzyl-5-chloro[1,2,3,6]tetrahydropyrid-3-one (5, R = Bn, Y = Cl) (0.51 g, 46%) as a yellow oil, i.r. (film)/cm-1 1685, 1610, 1295, 1110, 1030, 745, 700; p.m.r. (CDCl3) 3,23 (2H, br s), 3.49 (2H, br s), 3.71 (2H, s), 6.29 (1H, t, J 1.5Hz), 7.26-7.47 (5H, m); m/z (EI) 221 (M+), 130, 91 (100%).
5-Hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OH).
A cooled solution of sodium (8.6 g, 375 mmol) in methanol (75 ml) was diluted with ether (175 ml) and N-(2-hydroxypropyl)-N-methylglycine methyl ester (59.5 g, 375 mmol) added dropwise over 30 min. The mixture was stirred at room temperature for 2 h. before being treated with trifluoroacetic acid (30 ml, 390 mmol). The precipitate was removed by filtration, washed with methanol and dried in vacuo to give 5-hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OH) (27.1 g, 57%) as an amorphous solid m.p. >300°C. i.r. (Nujol)/cm-1 3400, 2790, 1900, 1605, 1570, 1460, 1320, 1180, 830; p.m.r. (200 MHz, D2O) 3.03 (3H, s), 3.83 (4H, s).
5-Methanesulfonyloxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2Me).
1,8-Diazabicyclo[5,4,0]undec-7-ene (1.6 ml, 10 mmol) was added to a suspension of 5-hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (1.27 g, 10 mmol) in anhydrous dichloromethane (5 ml) at room temperature. After 30 min. the solution was cooled to &endash;78°C and methanesulfonyl chloride (0.8 ml, 10 mmol) was added dropwise. The temperature was maintained for 30 min. then the mixture was allowed to warm to ambient temperature and the picture poured into ice/water (50 ml). The organic layer was rapidly separated and the aqueous layer extracted with dichloromethane. The combined organic extracts were dried (MgSO4), concentrated in vacuo to give methanesulfonyloxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2Me) (1.82 g, 89%) as an unstable yellow oil, i.r. (film)/cm-1 1690, 1645, 1375, 1185, 1140, 1115, 930, 795; p.m.r. (CDCl3)2.45 (3H, s), 3.15 (2H, t, J 0.7Hz), 3.26 (3H, s), 3.40 (2H, t, J 1.3Hz), 6.14 (1H, t, J 1.3Hz); c.m.r (CDCl3) 38.9, 44.2 55.0, 61.8, 113.7, 165.1, 195.2; m/z (EI) 205 (M+), 147, 126, 55 (100%).
1-Methyl-5-trifluoromethanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2CF3).
1,8-Diazabicyclo[5,4,0]undec-7-ene (0.7 ml, 5 mmol) was added to a suspension of 5-hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (0.635 g, 5 mmol) in anhydrous dichloromethane (5 ml) at room temperature. After 30 min. the solution was cooled to &endash;78°C and trifluoromethanesulfonic anhydride (0.8 ml, 10 mmol) was added dropwise. The temperature was maintained for 30 min. then the mixture was allowed to warm to ambient temperature and the solvent removed in vacuo. The brown residue was shaken vigorously with hexane (20 ml) and the hexane layer separated and evaporated to dryness to give 1-methyl-5-trifluoromethanesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2CF3) (0.35 g, 31%) as an unstable yellow oil which darkened rapidly on standing; i.r. (film)/cm-1 2975, 2790, 1700, 1665, 1430, 1145, 800; p.m.r. (200 MHz, CDCl3) 2.41 (3H, s), 3.19 (2H, br s,), 3.48 (2H, br s), 6.16 (1H, t, J 0.7Hz), m/z (EI) 259 (M+), 216, 126, 98, 55 (100%).
1-Methyl-5-toluenesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2C6H4Me).
1,8-Diazabicyclo[5,4,0]undec-7-ene (1.6 ml, 10 mmol) was added to a suspension of 5-hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (1.27 g, 10 mmol) in anhydrous dichloromethane (25 ml) at room temperature. The mixture was stirred for 1 h., then poured into cold water and extracted with dichloromethane (2 x 50 ml). The organic layers were combined, dried (MgSO4) and evaporated in vacuo to give 1-methyl-5-toluenesulfonyloxy[1,2,3,6]tetrahydropyrid-3-one (5, R = Me, Y = OSO2C6H4Me) (0.85 g, 42%) as an unstable orange oil, i.r. (film)/cm-1 1680, 1645, 1385, 1200, 1180, 1085, 750; p.m.r. (CDCl3) 2.41 (3H, s), 2.45 (3H, s), 3.07 (2H, br s), 3.28 (2H, br s), 5.89 (1H, br s), 7.39 (2H, d, J 8Hz), 7.38 (2H, d, J 8Hz).
5-Chloro-1-methyl[1,2,3,6]tetrahydropyridin-3-one (5, R = Me, Y = Cl).
1,8-Diazabicyclo[5,4,0]undec-7-ene (1.6 ml, 10mmol) was added to a suspension of 5-hydroxy-1-methyl[1,2,3,6]tetrahydropyrid-3-one (1.27 g, 10 mmol) in anhydrous dichloromethane (10 ml) at room temperature. After 30 min. the solution was cooled to 0°C and oxalyl chloride (1 ml, 11.5 mmol) added dropwise. The mixture wasstirred at this temperature for 1 h, poured into saturated aqueous potassium carbonate (20 ml) and extracted with diisopropyl ether( 2 x 25 ml). Solvent removal in vacuo afforded 5-chloro-1-methyl[1,2,3,6]tetrahydropyridin-3-one (5, R = Me, Y = Cl) (0.14 g, 9%) as an unstable yellow oil; p.m.r. (CDCl3) 2.54 (3H, s), 3.25 (2H, s), 3.46 (2H, s), 6.29 (1H, t, J 1.5 Hz); m/z (EI) 145/147 (M+), 102/104, 67.
4-(Cyclohex-2-en-1-on-3-yl)-1-triisopropylindole (9)and 4-(cyclohex-2-en-1-on-3-yl)-indole (10).
Butyllithium (1.6 M solution in hexanes, 0.7 ml, 1.25 mmol) was added dropwise to a solution of eta6-(1-triisopropylsilylindole)tricarbonylchromium(0) (0.51 g, 1.25 mmol) in anhydrous THF (10 ml) at &endash;78°C. The mixture was stirred at this temperature for 2 h. then transferred via a cannula to a suspension of copper bromide-dimethyl sulphide complex (0.52 g, 2.5 mmol) in THF (10 ml) at -40°C and the solution stirred for a further 1 h. Concurrently, 3-chlorocyclohex-2-en-1-one (0.3 g, 2.3 mmol)23 and tetrakistriphenylphosphinepalladium(0) (0.06 g, 5 mol %) were heated in THF (10 ml) under reflux for 30 min. and after cooling, the resulting solution was added to the indole cuprate via a syringe. The mixture was stirred for 1 h. then gradually allowed to warm to room temperature. The solution was opened to atmospheric oxygen and stirred at room temperature overnight. The resulting suspension was filtered through a pad of Celite and the filtrate poured into aqueous ammonium chloride (15%w/v, 50 ml). The aqueous solution was extracted with ether (30 ml) and the extract dried (MgSO4) and concentrated in vacuo . The crude product was purified by radial chromatography (ether: petroleum ether 0:100 &endash;> 80:20) to give 4-(cyclohex-2-en-1-on-3-yl)-1-triisopropylindole (9) (0.672 g, 36%) as a yellow oil, i.r. (film)/cm-1 1645, 1605, 1460, 1415, 1280, 1165, 1135, 765; p.m.r. (CDCl3) 1.15 (18H, d, J 7.5Hz), 1.71 (3H, sept, J 7.5Hz), 2.20 (2H, quintet, J 6Hz), 2.55 (2H, t, J 6Hz), 2.90 (2H, br t, J 6Hz), 6.54 (1H, t, J 1Hz), 6.79 (1H, dd, J 2, 0.5Hz), 7.16-7.18 (2H, m), 7.33 (1H, d, J 2Hz), 7.51-7.56 (1H, m); c.m.r. (CDCl3) 12.87, 18.13, 23.38, 30.12, 37.6, 104.1, 115.2, 118.6, 121.2, 127.4, 128.7, 132.3, 141.3, 161.9, 200.2; m/z (EI) 367 (M+)324, (100%), 210. Found: M+ 367.2332; C23H33NOSi requires: 367.2331.
Subsequently eluted was 4-(cyclohex-2-en-1-on-3-yl)-indole (10) (0.156 g, 14%) as a yellow oil, p.m.r. (CDCl3)2.24 (2H, quintet, J 6Hz), 2.90 (2H, br t, J 5Hz0, 6.56 (1H, t, J 1.5Hz), 6.72 (1H, m), 6.94 (1H, dd, J 8, 1Hz), 7.17-7.38 (2H, m), 7.42-7.45 (1H, m), 8.43 (1H, br s); m/z (EI) 211 (M+), 183, 154 (100%). Found: M+ 211.0989. C14H13NO requires: 211.0997.
4-(1-Benzyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)indole (11, P = H).
Butyllithium (1.6M solution in hexanes, 1.9 ml, 3 mmol) was added dropwise to a solution of eta6-(1-triisopropylsilylindole)tricarbonylchromium(0) (0.51 g, 3 mmol) in anhydrous THF (10 ml) at &endash;78°C. The mixture was stirred at this temperature for 2 h, transferred via a cannula to a suspension of copper bromide-dimethyl sulfide complex (0.52 g) in THF (10 ml) at &endash;40°C and stirred for a further 1h. Concurrently, 1-benzyl-5-chloro[1,2,3,6]tetrahydropyrid-3-one (1.1g, 5 mmol) and tetrakistriphenylphosphinepalladium(0) (0.06g) were heated in THF (10 ml) under reflux for 30 min.and, after cooling, were addedto the indole cuprate via a syringe. The mixture was stirred for 1 h, gradually warmed to room temperature then heated to 60°C for 30 min. The mixture was decomplexed and worked up as above. Radial chromatography as above gave 4-(1-benzyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)indole (11) (0.41 g, 45%) as a yellow oil, i.r. (film)/cm-1 3240, 1665, 1600, 1465, 1135, 900, 765; p.m.r. (CDCl3) 3.32 (2H, br s), 3.78 (2H, s), 3.79 (2H, s), 6.59 (1H, t, J 1.5Hz), 6.64 (1H, m), 7.11 (1H, dd, J 7.7, 1.5Hz), 7.18 (1H, t, J 7.7Hz), 7.27 (1H, dd, J 3.5, 6Hz), 7.31-7.38 (5H, m), 7.42 (1H, dt, J 7.7, 1Hz), 8.5 (1H, br s); c.m.r. (CDCl3) 55.2, 60.9, 61.8, 102.3, 113.1, 118.8, 121.9, 125.4, 127.7, 128.1, 128.6, 128.7, 129.2, 129.8, 136.4, 136.7, 159.9, 196.6; m/z (CI) 303 (M+H+), 211, 183, 91 (100%). Found (EI): M+, 302.1409; C20H18N20 requires 302.1419.
Also eluted was an inseparable mixture of 4-(1-benzyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole , (see below) p.m.r. (CDCl3) inter alia 1.14 (18H, d, J 7.5Hz), 1.70 (3H, sept, J 7.5Hz), 3.16 (2H, s), 3.30 (2H, s), 3.78 (2H, s), 6.75 (1H, dd, J 4, 1Hz), 7.15-7.20 (3H, m), 7.50-7.62 (1H, m) and 1-benzyl-5-butyl[1,2,3,6]tetrahydrpyrid-3-one, p.m.r. (CDCl3) 0.9 (3H, t, J 6.9Hz), 1.13-1.6 (4H, m), 2.20 (2H, t, J 7Hz), 3.12 (4H, m), 3.65 (2H, s),5.94 (1H, br s), 7.25-7.45 (5H, m).
4-(1-Benzyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (11, P = SiiPr3)
Butyllithium (1.5M in hexanes, 1.46 ml, 2.1 mmol) was added dropwise to a solution of 4-bromo-1-triisopropylindole (0.77 g, 2.2 mmol) in THF (10 ml) at &endash;78°C. After 30 min, the lithiated compound was transferred, via a cannula, into a flask containing a suspension of copper bromide-dimethyl sulfide complex (0.75 g, 3.6 mmol) in THF (10 ml) at &endash;40°C. The dark mixture was stirred at this temperature for a further 60 min. Concurrently, 1-benzyl-5-chloro[1,2,3,6]tetrahydropyrid-3-one (0.45g, 2.2 mmol) and tetrakistriphenylphosphinepalladium(0) (0.05 g) were refluxed in THF (10 ml) for 30 min. The cooled catalyst mixture was added to the organocuprate at &endash;40°C and the mixture allowed to warm to room temperature over 10 min. After a further 1 h, the reaction was quenched with aqueous ammonium chloride (15% w/v, 50 ml) and the organic components extracted with ether (3 x 50 ml). The combined organic phases were washed with water (50 ml) and brine (50 ml), dried (MgSO4) and the solvent removed in vacuo . Column chromatography of the crude product (SiO2, ether:hexane 1:3) gave 4-(1-benzyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (11, P = SiiPr3) (0.45 g, 45%) as a yellow semisolid, i.r. (Nujol)/cm-1 1670, 1610, 1425, 1280, 1155, 1020, 880, 760; p.m.r. (CDCl3) 1.14 (18H, d, J 7.5Hz), 1.70 (3H, sept, J 7.4Hz), 3.31 (2H, br s), 3.80 (4H, s), 6.60 (1H, br s), 6.74 (1H, d, J 1Hz), 7.10-7.15 (2H, m), 7.30-7.35 (6H, m), 7.55 (1H, br d, J 7.5Hz); m/z (EI) 457 (M+), 367, 323, 295, 183, 91 (100%). Found: M+, 458.2745; C29H38N2OSi requires: 458.2753.
4-(1-Methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (4, P = SiiPr3).
Method 1 : In an analogous manner to above, 4-bromo-1-triisopropylindole (1.20 g, 3.6 mmol) was lithiated at &endash;78°C and coupled with 5-chloro-1-methyl[1,2,3,6]tetrahydropyrid-3-one (0.54 g, 3.6 mmol). Work up as above and column chromatography (SiO2, methanol:dichloromethane 2:98) of the crude product gave 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (4, P = SiiPr3) (0.39 g, 31%) as yellow crystals m.p. 102-104°C, i.r. (Nujol)/cm-1 1675, 1420, 1270, 1155, 1130, 880, 760; p.m.r. (400 MHz, CDCl3) 1.15 (18H, d, J 7.5Hz), 1.71 (3H, d, J 7.4Hz), 2.53 (3H, s), 3.26 (2H, br s), 3.73 (2H, br s), 6.59 (1H, t, J 1.5Hz), 6.78 (1H, dd, J 3, 0.8Hz), 7.14-7.19 (2H, m), 7.33 (1H, d, J 3Hz), 7.54-7.57 (1H, m); c.m.r. (CDCl3) 12.94, 18.19, 45.12, 57.58, 63.05, 104.3, 115.7, 118.7, 121.2, 125.2, 128.9, 129.8, 132.3, 141.5, 160.2, 196.4; m/z (EI)382 (M+), 339, 296, 154, (100%).
Method 2 : Tris(dibenzylideneacetone)dipalladium(0)chloroform complex (0.1 g, 6mol %) was added to a mixture of 1-triisopropylsilylindole-4-boronic acid (0.05g, 0.15 mmol), 1-methyl-5-trifluoromethanesulfonyloxy[1,2,3,6]tetrahydropyridin-3-one (0.30 g, 1.1 mmol) and caesium fluoride (0.5 g, 3.3 mmol) in 1,2-dimethoxyethane (10 ml0 and the mixture refluxed for 1 h. The solution was directly chromatographed (SiO2; hexane:ethyl acetate 3:2) to give 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (4, P = SiiPr3) (0.035 g, 60%) as a yellow oil, spectroscopically identical to the material obtained above.
Method 3 : 5-Methanesulfonyloxy-1-methyl[1,2,3,6]tetrahydropyridin-3-one (1.50 g, 5 mmol) was added to a mixture of lithium chloride (1.0 g, 24 mmol), lithium carbonate (2.0 g, 27 mmol) and tris(dibenzylideneacetone)dipalladium(0)chloroform complex (0.15 g, 2 mol %) in 1,2-dimethoxyethane (10 ml)at room temperature. Whem the colour of the catalyst mixture had changed from purple to yellow, a solution of 1-triisopropylsilyl-4-trimethylstannylindole (2.5 g, 6 mmol) in DME (10 ml) was added in one portion and the mixture heated at 60°C. The reaction was monitored by reverse phase C18 tlc analysis (eluent; DCM:acetonitrile 1:3) and upon completion (1 h), the mixture was cooled and filtered through a Celite pad and the filtrate evaporated to dryness. The crude residue was partitioned between water (50 ml) and chloroform (50 ml) and the organic layer separated, washed with water (5 x 50 ml), dried (MgSO4) and evaporated to dryness. The residue was purified by column chromatography (SiO2, ethyl acetate:hexane 1:1) to give 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (4, P = SiiPr3) (0.88 g, 38%) asa yellow oil, spectroscopically identical to the material obtained before.
4-(1-Methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)indole (4, P = H).
Tetrabutylammonium fluoride (1.1 M in THF, 0.5 ml, 0.55 mmol) was added dropwise to a solution of 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (0.13 g, 0.35 mmol) in dichloromethane (10 ml) at room temperature. Water (10 ml) was added after 5 min. and the organic layer separated. The aqueous layer was washed with dichloromethane (2 x 10 ml) and the combined organic phases washed with water (10 ml) and brine (10 ml), dried (MgSO4) and evaporated to dryness. Column chromatography of the crude product (SiO2, methanol: dichloromethane 2:98) gave 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)indole (4, P = H) (0.069 g, 84%) as a yellow oil, i.r. (film)/cm-1 3300, 2795, 1660, 1590, 1460, 1285, 900, 750; p.m.r. (CDCl3) 2.54 (3H, s), 3.26 (2H, br s), 3.74 (2H, br s), 6.62 (1H, t, J 1.5Hz), 6.72 (1H, m), 7.19-7.26 (2H, m), 7.31 (1H, t, J 7Hz), 7.38 (1H, m), 8.41 (1H, br s); m/z (EI) 226 (M+), 183, 154 (100%). Found: (M+&endash;CH2NMe) 183.0675; C12H9NO requires 183.0684.
4-(1-Methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)gramine (3, P = H, X = NMe2).
Tetrabutylammonium fluoride (1.0 M in THF, 0.2 ml) was added dropwise to a mixture of Eschenmoser's salt (0.2 g, 1.1mmol) and 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)-1-triisopropylsilylindole (0.39 g, 1.1 mmol) in dichloromethane (10 ml) at room temperature. After 3 h. water (25 ml) was added and the mixture extracted withdichloromethane (2 x 25 ml). The organic phase was washed with water (3 x 50 ml) and brine (50 ml), dried (MgSO4) and evaporated to dryness. The residual oil was purified by chromatography (Al2O3, ether) to give 4-(1-methyl[1,2,3,6]tetrahydropyrid-3-on-5-yl)gramine (3, P = H, X = NMe2) (0.08 g, 22%) as a yellow oil, i.r. (film)/cm-1 1670, 1430, 1265, 1140, 1050, 760; p.m.r. (CDCl3) 2.23 (6H, s), 2.44 (3H, s), 3.17 (2H, br s), 3.64 (2H, br s), 4.67 (2H, s), 6.52 (1H, t, J 1.5Hz), 6.58 (1H, dd, J 3, 0.8Hz), 7.06-7.15 (2H, m), 7.44 (1H, br t, J 8Hz); m/z (EI) 283 (M+), 58 (100%).
References
1. P.A. Stadler and P. Stutz in 'The Alkaloids', ed. R.H.F. Manske, Academic Press, New York, 1975, p.1.
2. E.C. Kornfeld, E.J. Fornefeld, G.B. Kline, M.J. Mann,D.E. morrison, R.G. Jones and R.B. Woodward, J. Am. Chem. Soc., 1956, 78, 3087.
3. G.W. Gribble, Contemporary Org. Syn., 1995, 1, 145.
4. See for example: A.P. Kozikowski, K. Sato, A. Basu and J.S. Lazo, Liebigs Ann. Chem., 1989, 111, 6228.
5. K. Teranishi, S. Hayashi, S.-I. Nakatsuka and T. Goto, Synthesis , 1995, 506 and references there cited.
6. M. Somei, K. Kizu, M. Kunimoto and F. Yamada, Chem. Pharm. Bull., 1985, 33, 3696.
7. R.M. Moriarty, Y. Ku and U.S. Gill. Organometallics, 1988, 7, 660.
8. D.A. Widdowson, Phil. Trans. R. Soc. Lond., 1988, 326 A, 595; M.F. Semmelhack, G.R. Clark, J.L. Garcia, J.J. Harrison, Y. Thebtaranonth, W. Wulff and A. Yamashita, Tetrahedron, 1981, 37, 3957.
9. A.P. Kozikowski and K. Isobe, J. Chem. Soc., Chem. Commun., 1978, 1076.
10. G. Jaouen, Ann. N. Y. Acad. Sci., 1977, 295, 59; M.F. Semmelhack, Ann. N. Y. Acad. Sci., 1977, 295, 32.
11. P.J. Beswick, C.S. Greenwood, T.J. Mowlem, G. Nechvatal and D.A. Widdowson, Tetrahedron, 1988, 44, 7325.
12. M.J. Dickens, PhD Thesis, London University, 1989.
13. D.M.A. Conlon, PhD Thesis, London university, 1996.
14. M.P. Moyer, J.F. Shiurba and H. Rapoport, J. Org. Chem., 1988, 51, 5106.
15. J.E. McMurry, Acc. Chem. Res., 1974, 7, 281.
16. N. Masters and D.A. Widdowson, Chem. Soc. Chem. Commun., 1983, 955.
17. John Wyeth, Brit. Pat. No. GB-2060617.
18. C.J. Kowalski and K.W. fields, J. Org. Chem., 1981, 46, 197.
19. R.F. Heck, 'Palladium Reagents in Organic Synthesis', Academic Press, London, 1985, 179 et seq.
20. A. Suzuki, Pure Appl. Chem., 1991, 63, 419.
21. J. Schreiber, H. maag, N. Hashimoto and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1971, 10, 330.
22. S. Raucher and G.A. Koolpe. J. Org. Chem., 1986, 48, 2066.
23. R.E. Mershaw, Tetrahedron Lett., 1989, 30, 3573.