| [Related articles/posters: 087 026 092 ] |
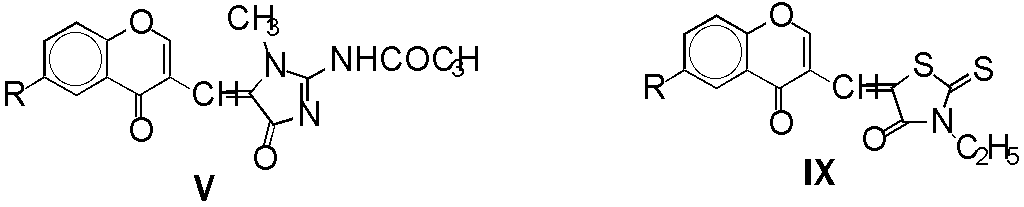
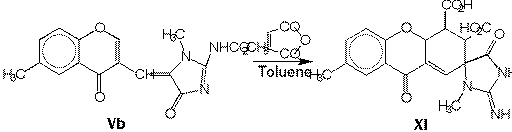
Compound |
R |
Formula |
m.p. |
wi (calcd)/ % wI (found)/ % |
Yield |
tr | ||
Mr |
°C |
C |
H |
N |
% |
min | ||
Va |
H |
C16H13N3O4 |
246 - 248 |
61.73 |
4.21 |
13.50 |
75 |
3 |
311.29 |
61.31 |
4.14 |
13.41 |
71 |
60 | |||
Vb |
CH3 |
C17H15N3O4 |
277 - 279 |
62.76 |
4.65 |
12.92 |
60 |
4 |
325.32 |
62.15 |
4.66 |
12.59 |
57 |
120 | |||
Vc |
Cl |
C16H12ClN3O4 |
268 - 270 |
55.58 |
3.50 |
12.15 |
76 |
2 |
345.74 |
55.58 |
3.59 |
12.13 |
72 |
60 | |||
Vd |
Br |
C16H12BrN3O4 |
270 - 272 |
49.25 |
3.10 |
10.77 |
84 |
1 |
390.19 |
48.91 |
3.02 |
10.54 |
- |
- | |||
Ve |
AcO |
C18H15N3O6 |
259 - 262 |
58.54 |
4.09 |
11.38 |
46 |
3 |
369.33 |
58.06 |
4.07 |
11.28 |
40 |
90 | |||
Vf |
NO2 |
C16H12N4O6 |
285 - 286 |
53.94 |
3.39 |
15.72 |
84 |
3 |
356.29 |
53.83 |
3.29 |
15.43 |
- |
- | |||
VIa |
H |
C14H11N3O3 |
250 - 252 |
62.45 |
4.12 |
15.61 |
70 |
3 |
269.26 |
62.38 |
3.96 |
15.72 |
| ||||
VIb |
CH3 |
C15H13N3O3 |
294 - 297 |
63.60 |
4.63 |
14.83 |
67 |
3 |
283.28 |
63.24 |
4.68 |
14.06 |
|||||
VIc |
Cl |
C14H10ClN3O3 |
356 - 360 |
55.36 |
3.32 |
13.83 |
92 |
3 |
303.7 |
55.21 |
3.49 |
13.22 |
|||||
VId |
Br |
C14H10BrN3O3 |
276 - 278 |
48.30 |
2.89 |
12.07 |
68 |
3 |
348.16 |
48.25 |
2.96 |
12.44 |
|||||
VIe |
NO2 |
C14H10N4O5 |
237 - 240 |
53.51 |
3.21 |
17.83 |
98 |
3 |
314.26 |
52.94 |
2.99 |
17.39 |
|||||
VIIa |
H |
C15H11N3O5 |
253 - 255 |
57.82 |
3.51 |
13.41 |
69 |
6 |
313.3 |
57.50 |
3.90 |
13.08 |
| ||||
VIIb |
Cl |
C15H10ClN3O5 |
356 - 360 |
54.34 |
3.76 |
11.18 |
71 |
4 |
375.76 |
54.69 |
3.23 |
11.23 |
Table 1. Continued.
Compound |
R |
Formula |
m.p. |
wi (calcd)/ % wI (found)/ % |
Yield |
tr | ||
Mr |
°C |
C |
H |
N |
% |
min | ||
VIIc |
CH3 |
C16H13N3O5 |
299 - 301 |
58.71 |
3.97 |
12.84 |
58 |
7 |
327.3 |
58.44 |
4.30 |
12.75 |
|||||
VIId |
Br |
C15H10BrN3O5 |
350 |
45.92 |
2.55 |
10.71 |
60 |
7 |
392.2 |
46.28 |
2.36 |
10.48 |
|||||
VIIe |
NO2 |
C15H10N4O7 |
320 - 321 |
50.25 |
2.79 |
15.63 |
60 |
2 |
358.2 |
49.92 |
4.09 |
15.78 |
|||||
VIIIa |
H |
C13H8N2O3S |
292 - 295 |
57.35 |
2.96 |
10.29 |
79 |
8 |
272.3 |
56.98 |
2.91 |
10.05 |
72 |
60 | |||
VIIIb |
CH3 |
C14H10N2O3S |
315 - 317 |
58.73 |
3.52 |
9.78 |
70 |
6 |
286.3 |
58.50 |
3.52 |
9.04 |
66 |
30 | |||
VIIIc |
Cl |
C13H7ClN2O3S |
319 - 321 |
50.91 |
2.30 |
9.13 |
74 |
4 |
306.7 |
51.03 |
2.34 |
9.05 |
71 |
30 | |||
VIIId |
Br |
C13H7BrN2O3S |
329 - 331 |
44.46 |
2.01 |
7.98 |
96 |
9 |
351.2 |
44.53 |
2.02 |
7.16 |
88 |
60 | |||
VIIIe |
AcO |
C15H10N2O5S |
303 - 305 |
54.54 |
3.05 |
8.48 |
62 |
10 |
330.3 |
53.82 |
2.98 |
7.99 |
59 |
120 | |||
IXa |
H |
C15H11NO3S2 |
215 - 217 |
56.77 |
3.49 |
4.41 |
70 |
5 |
317.4 |
57.07 |
3.48 |
4.45 |
74 |
60 | |||
IXb |
Cl |
C15H10ClNO3S2 |
231 - 233 |
51.21 |
2.86 |
3.98 |
67 |
5 |
351.8 |
50.96 |
2.82 |
3.79 |
65 |
60 | |||
Xa |
H |
C14H15N5O5 |
325 - 327 |
55.08 |
4.95 |
13.76 |
52 |
|
305.3 |
decomp. |
55.35 |
4.59 |
13.65 |
||||
Xe |
OH |
C14H15N3O6 |
315 - 317 |
52.33 |
4.70 |
13.08 |
47 |
|
321.3 |
decomp. |
52.12 |
4.59 |
12.72 |
||||
Xf |
NO2 |
C14H14N4O7 |
>360 |
48.00 |
4.02 |
15.98 |
56 |
|
350.3 |
decomp. |
48.09 |
3.855 |
15.77 |
*
The upper data of yield and reaction time (tr) are
given for the condensation in microwave oven, the lower data for the classic
condensation.
Table 2. IR Spectral Data of Synthesized Compounds
[nu]/cm-1 | ||||
Compoud |
[nu](NH) |
[nu](C=O)heterocycl. |
[nu](C=O)pyrone |
[nu](C=O)other |
Va |
3078 - 3162 |
1739 |
1650 |
1643 |
Vb |
3070 - 3175 |
1740 |
1650 |
1642 |
Vc |
3075 - 3179 |
1735 |
1659 |
1636 |
Vd |
3075 - 3180 |
1729 |
1658 |
1640 |
Ve |
3081 - 3181 |
1739 |
1652 |
1630, 1758 |
Vf |
3080 - 3177 |
1745 |
1660 |
1641 |
VIa |
3070 - 3185 |
1720 |
1650 |
|
VIb |
3095 - 3180 |
1720 |
1645 |
|
VIc |
3070 - 3175 |
1735 |
1655 |
|
VId |
3090 - 3180 |
1721 |
1655 |
|
VIe |
3094 - 3186 |
1719 |
1658 |
|
VIIa |
3080 - 3230 |
1719 |
1657 |
1740 |
VIIb |
3075 - 3225 |
1721 |
1658 |
1742 |
VIIIa |
3038 - 3180 |
1740 |
1665 |
|
VIIIb |
3086 - 3185 |
1739 |
1668 |
|
VIIIc |
3070 - 3190 |
1742 |
1665 |
|
VIIId |
3060 - 3180 |
1736 |
1668 |
|
VIIIe |
3078 - 3185 |
1745 |
1663 |
1750 |
IXa |
3069 - 3110 |
1688 |
1660 |
|
IXb |
3060 - 3110 |
1688 |
1662 |
Table 2 Continued
[nu]/cm-1 |
|||||
Compound |
[nu](OH) |
[nu](NH) |
[nu](C0) |
[nu](CO)acid |
[nu](C=C) |
Xa |
3480-3400 |
3080-3040 |
1740 |
1700 |
1646 |
Xe |
3540-3440 |
3080-3020 |
1748 |
1700 |
1642 |
Xf |
3560-3480 |
3110-3010 |
1723 |
1700 |
1660 |
XI |
3460 |
3190-3120 |
1647a |
1720-1722 |
1620 |
- |
- |
1708b |
- |
- |
a[nu](CO)pyr,
b[nu](CO)het
Table 3. 1H NMR spectra data of prepared compounds
Compound |
Solvent |
1H NMR spectrum [delta] (ppm) |
Va |
CDCl3 |
2.25 (s, 3H, CH3); 3.39 (s, 3H, CH3-N); 6.86 - 8.32 (m, 5H, H-Ar); |
9.68 (s, 1H, H-2); 10.84 (s, 1H, NH). | ||
Vb |
DMSO |
2.50 (s, 3H, CH3); 2.54(s, 3H, CH3); 3.57(s, 3H, CH3-N); 6.56 (s, |
1H, H-9); 7.60 - 7.70 (m, 5H, H-Ar); 7.97 (s, 1H, H-5); 9.56 (s, 1H, | ||
H-2). | ||
Vc |
DMSO |
2.76 (s, 3H, CH3); 3.52 (s, 3H, CH3-N); 6.72 (s, 1H, H-9); 8.02 (d, |
1H, H-8, 3J=9Hz); 8.11 (d, 1H, H-7, 3J=9Hz); 8.32 (d, 1H, H-5, | ||
4J=2Hz); 9.54 (s, 1H, H-2). | ||
Vd |
DMSO |
2.78 (s, 3H, CH3); 3.53 (s, 3H, CH3-N); 6.74 (s, 1H, H-9); 7.95 (d, |
1H, H-8, 3J=9Hz); 8.24 (d, 1H. H-7, 3J=9Hz); 8.49 (d, 1H, H-5, | ||
4J=1.8Hz), 9.55 (s, 1H, H-2). | ||
Ve |
DMSO |
2.13 (s, 3H, CH3O); 2.3 (s, 3H, CH3O); 3.45 (s, 3H, CH3-N); 6.47 |
(s, 1H, H-9); 7.6 - 7.85 (m, 3H, H-8, 7, 5); 9.30 (s, 1H, H-2); 11.41 | ||
(s (broad), 1H, N-H). | ||
Vf |
DMSO |
2.13 (s, 3H, CH3O); 3.21 (s, 3H, CH3-N); 6.43 (s, 1H, H-9); 8.00(s, |
1H, H-8); 8.64 (s, 1H, H-7); 8.85 (s, 1H, H-7); 8.85 (s, 1H, H-5); | ||
9.353 (s, 1H, H-2). | ||
VIa |
DMSO |
3.35 (s, 3H, CH3-N); 6.24 (s, 1H, H-9); 7.52 (dd, 1H, H- |
7,3J=7.8Hz); 7.69 (dd, 1H, H-8, 3J=7.8Hz); 7.81 (dd, 1H, H-6, | ||
3J=7.8Hz); 8.12 (dd, 1H, H-5, 3J=7.8Hz); 9.88 (s, 1H, H-2); 7.4 - | ||
8.4 (broad, 1H, NH). |
Table 3. Continued
Compound |
Solvent |
1H NMR spectrum [delta] (ppm) |
VIb |
DMSO |
2.45 (s, 3H, CH3); 3.38 (s, 3H, CH3N); 6.25 (s, 1H, H-9); 7.60 - |
7.95 (m, 3H, H-8, 7, 5); 9.86 (s, 1H, H-2); 7.4 - 8.4 (broad, 1H, | ||
NH). | ||
VIIa |
DMSO |
3.39 (s, 3H, CH3CN); 6.19 (s, 1H, H-9); 7.53 (dd, 1H, H-6, |
3J=7Hz); 7.69 (d, 1H, H-8, 3J=7Hz); 7.84 (dd, 1H, H-7, 3J=7Hz); | ||
8.12 (d, 1H, H-5, 3J=7Hz); 9.8 (s, 1H, H-2). | ||
VIId |
DMSO |
3.39 (s, 3H, CH3CN); 6.19 (s, 1H, H-9); 7.70 (d, 1H, H-8, 3J=8Hz); |
7.97 (dd, 1H, H-7, 3J=8Hz, 4J=2.2Hz); 8.17 (d, 1H, H-5, 4J=2.2Hz); | ||
9.895 (s, 1H, H-2). | ||
VIIe |
DMSO |
3.44 (s, 3H, CH3-N); 6.59 (s, 1H, H-9); 8.00 (d, 1H, H-8, 3J=9Hz); |
8.61 (dd, 1H, H-7, 3J=9Hz, 4J=2.5Hz); 8.79 (d, 1H, H-5, | ||
4J=2.54Hz); 9.395 (s, 1H, H-2); 9.64 (s( broad), 1H, NH). | ||
VIIIc |
DMSO |
6.36 (s, 1H, H - 9); 7.78 (d, 1H, H-8, 3J=10Hz); 7.87 (dd, 1H, H - 7, |
3J=10Hz, 4J=2.2Hz); 8.23 (d, 1H, 4J=2.2Hz); 8.94 (s, 1H, H-5); | ||
11.65 (s, 1H, NH); 12.46 (s, 1H, OH). | ||
VIIId |
DMSO |
6.35 (s, 1H, H - 9); 7.74 (d, 1H, H-8, 3J=8.8Hz); 8.02 (dd, 1H, H - 7, |
3J=8.8Hz, 4J=2.2Hz); 8.22 (d, 1H, 4J=2.2Hz); 8.92 (s, 1H, H-2); | ||
VIIIe |
DMSO |
2.33 (s, 3H, CH3); 6.38 (s, 1H, H - 9); 7.60 (dd, 1H, H-7, 3J=9Hz, |
4J=1.9Hz); 7.83 (d, 1H, H-8, 3J=9Hz); 7.88 (d, 1H, H-5, 4J=1.9Hz); | ||
8.94 (s, 1H, H-2); 11.80 (s, 1H, NH); 12.45 (s, 1H, OH). |
Table 3. Continued
Compound |
Solvent |
1H NMR spectrum [delta] (ppm) |
IXb |
DMSO |
2.490 (t, 3H, CH3); 5.346 (q, 2H, CH2); 8.84 (s, 1h, H-2); 9.07 (d, |
1H, H-8, 3J=9.1Hz); 9.072 (dd, 1H, H-7, 3J=9.1Hz, 4J=2Hz); 9.36 | ||
(d, 1H, H-5, 4J=2Hz); 10.27 (s, 1H, H-9). | ||
Xa |
DMSO |
6.27 (s, 1H, CH); 7.23 - 7.66 (m, H, Ar - H); 9.25 (s, 1H, CH); |
10.26 (b., 6H, NH2, NH, OH). | ||
Xe |
DMSO |
3.38 (s, 3H, CH3); 6.70 (s, 1H, CH); 7.29 - 7.39 (dd, 1H, H-5, |
J=9Hz); 7.39 - 7.41 (d, 1H, H-3, J=3Hz); 7.57 - 7.60 (d, 1H, H-6, | ||
J=9Hz); 9.23 (s, 1H, CH); 10.28 (b., 7H, OH, NH, NH2). | ||
Xf |
DMSO |
3.68 (s, 3H, CH3); 7.95 - 8.81 (m, 3H, Ar-H); 9.32 (s, 1H, CH); |
10.00 (b., 6H, OH, NH, NH2). | ||
XI |
DMSO |
2.46 (s, 3H, CH3); 2.48 (s, 3H, CH3); 4.71 (broad, 5H, NH amide, |
CO2H, CH); 6.23 - 6.56 (m, 4H, Ar-H, H-2); 7.64 (s, 1H, =CH); | ||
9.47 (s, 1H, NH-imine). |
Results and Discussion
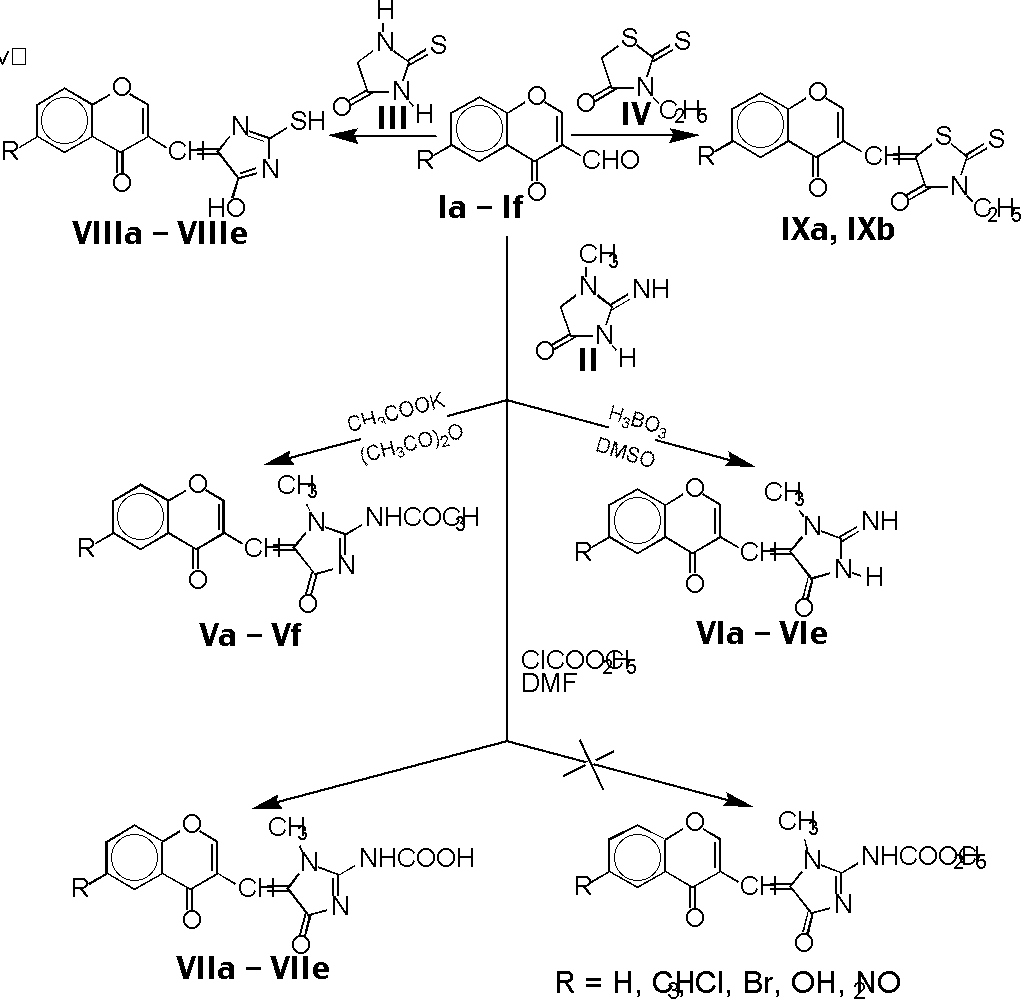
2-Acetamido-1-methyl-5-[(6-R-4-oxo-4H-benzopyran-3-yl)methylidene]-4,5-dihydroimidazol-4-ones
Va - Vf were obtained by condensations of Ia - If
with creatinine II were carried out in acetic anhydride, both under
classical and microwave irradiation conditions.
Although the yields in both methods were almost the same (46 - 84 %), reactions
in microwave oven were considerably shorter (Table 1).
When boric acid instead of potassium acetate was used as a catalyst and
reaction was carried out in dimethyl sulfoxide,
2-imino-1-methyl-5-[(6-R-4-oxo-4H-[1]-benzopyran-3-yl)
methylidene]imidazolidin-4-ones VIa - VIe were obtained in 67 -
98 % yields.
A convenient synthesis of
1-metyl-4-oxo-5-[(6-R-4-oxo-4H-[1]-benzopyran-3-yl)methylidene]-4,5-dihydroimidazol-2-carbamoic
acids VII was realized by reaction of creatinine II with
ethylchloroformate in N, N - dimethylformamide with subsequent addition of
aldehyde I. The hydrolyzed products VIIa - VIIe with 69 -
71 % yields were obtained by both classical and microwave irradiation methods,
respectively. Compounds VII resulted from utilization of condensing
water in the process of hydrolysis, because carbamoic acids VII have
been created in all our experiments, also in case of anhydrous conditions. In
the 1H NMR spectra of compounds VII did not occur signals for
ethyloxygroup.
Condensations of I with thiohydantoin III were
carried out in acetic anhydride in the presence of potassium acetate. The
difference of reaction times between these two condensation methods was also
evident although the yields of
2-thioxo-5-[(6-R-4-oxo-4H-[1]-benzopyran-3-yl)
methylidene]imidazolidin-4-ones VIIIa -VIIIe were comparable (59
- 96%).
Reactions of I with 3-ethylrhodanine IV in microwave oven
lead to arising of
2-thioxo-5-[(6-R-4-oxo-4H-benzopyran-3-yl)methylidene]thiazolidin-4-ones
IXa and IXb in 67 - 70 % yields.
All condensation products are stable solid compounds, rather insoluble in
common solvents, with high melting points. For their difficult solubility in
DMSO we had to make their 1H NMR spectra measured at elevated
temperature.
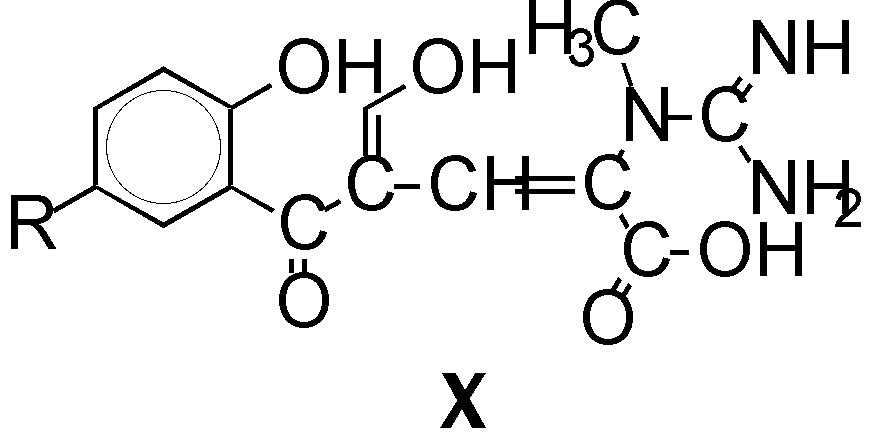
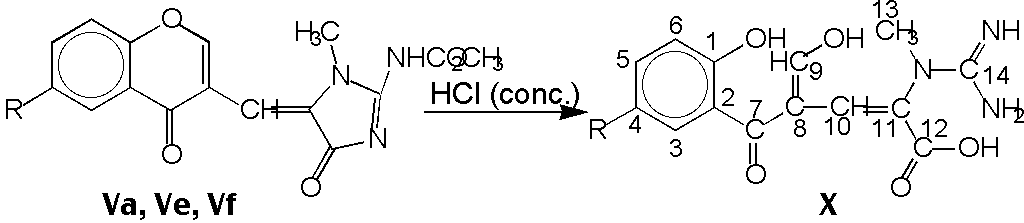
We tried to hydrolyze some of prepared compounds in diluted mineral acids, but
use of concentrated hydrochloric acid was successful only by case of 4 hrs.
reflux. Thus were isolated compounds X with both opened heterocycles
(yields 47 - 56 %). On the base of 1H NMR -, 13C NMR -
spectra and elemental analysis we could propose the structure of compounds
X, which contain guanidinyl, carboxyl and enolic groups. Compounds
X can be regarded as a mixtures of isomers with a very fast tautomeric
equilibrium both enolic and oxo groups. The assumption of fast tautomeric
equilibrium is supported by the high data for shift signals of C-9 in
13C NMR spectra.
We attempted to realize Diels - Alder reactions with various dienophiles,
using e.g. maleinanhydride, diethyl butindioate and tetracyanoethylene classic
as well as microwave irradiation - focused procedures. Only heating a mixture
of Vb in toluene with surplus of maleinanhydride at 40 oC
over 15 hrs. was successful and was formed out spiroheterocyclic aducts
XI (yield 64 %). The using of microwave irradiation for Diels - Alder
experiments was unsuccessful.
Summary
The synthesis of different types of 3-substituted 4H-4-oxobenzopyranes
was realized under focused microwave irradiation as well as by a classic
method.
The beneficial effect of microwave irradiation on aldol condensation of
3-formylchromones with 2-imino-1-methylimidazolidine-4-one (creatine),
2-thioxoimidazolidine-4-one (thiohydantoin) and
2-etyl-2-thioxothiazolidin-4-one (3-ethylrodanine) in different reaction media
is described. Our results showed that the effect of microwave irradiation on
studied reactions caused the shortening of reaction time, smoothly increased
the yields. The subsequent product reaction with some nucleophiles are
discussed. The structures of products has been prooved by elemental analysis,
IR - and 1H NMR - spectra.
References
1. Stankovièová, H., Fabian, W.M., Lácová, M.
Molecules 1, 223 - 235 (1996)
2. Stankovièová, H., Gasparová, R., Lácová,
M., Chovancová, J. Collect. Czech. Chem. Commun. 62, 781
(1997)
3. Lácová, M., Stankovièová, H., Odlerová,
Z. Il Pharmaco 50, 885 (1995)
4. El-Shaaer, H.M., Perjéssy, A., Zahradník, P.,
Lácová, M., Matulová, M. Monatsh. Chem. 124, 539
(1993)
5. Lácová, M., El-Shaaer, H.M., Loos, D., Matulová, M.,
Chovancová, J., Furdík, M. Molecules 3, 120 - 131
(1998)
6. Gasparová, R., Lácová, M., El-Shaaer, H.M.,
Odlerová, Z. Il Pharmaco 52, 251 - 253 (1997)
7. Lácová, M., Chovancová, J., Veverková, J., Toma,
S. Tetrahedron 52, 14995 - 15006 (1996)
8. Nohara, A., Sugihara, H., and Ukawa, K., Japan. Kokai Tokkyo Koho
78, 111,070 (1978); Chem. Abstr. 90, 54828z (1979).
9. Klutchko, S., Kaminski, D., and Von Strandtmann, M., U.S. Patent 4,008,252
(1977); Chem. Abstr. 87, 5808a (1977).
10. Lácová, M., El-Shaaer, H. M., Odlerová, Z., and
Furdík, M. Chem. Papers 48, 344 (1994).
11. Deulofeu, V., and Guerrero, T. J. Org. Synth. Coll. 3, 586
(1955).
12. Coirnwaite, W. R., Lazarus, S., Snelling, R. H., and Denoon, C. E. J.
Am. Chem. Soc. 58, 628 (1936).
13. Fitton, A. O., Frost, J. R., Suschitsky, H., and Houghton, P. G.
Synthesis 133 (1977).
14. Gasparová, R. and Lácová, M. Collect. Czech. Chem.
Commun. 60, 1178 (1995).
15. Villemin, D. and Martin, B. Synth. Commun. 25, 3135
(1995).
16. Villemin D., and Ricard M. Synth. Commun. 17, 283 (1987).
17. Nohara, A., Ishiguto, T., Sanno, Y. Tetrahedron Lett. 13, 1183
(1974)
Acknowledgments. The authors' thanks are due to Dr. K.
Gáplovská for elemental analyses. Mgr. I. Prokes for measurement
of proton NMR spectra and Dr. A. Perjéssy for IR spectra measurement,
members of Faculty of Natural Sciences, Comenius University, Bratislava.
Financial support for this research by the Slovak Grant Agency is gratefully
acknowledged, Grant No. 1/5085/98.