| [Related articles/posters: 091 011 102 ] |
Departamentos de Química Orgánicaa e Inorgánicab, Facultad de Química, Universidad de Murcia, Campus de Espinardo, E-30071, Murcia, Spain.
Control of self-organization at the molecular level is a field of major interest in molecular design and engineering. Molecular self-assembly, defined as the evolution towards spatial confinement through spontaneous connection of a few components, resulting in the formation of discrete entities at the molecular, covalent level,[1] is in fact a special type of synthetic procedure where several reactions between several reagents occur in one experimental operation to yield the final covalent structure. Molecular self-assembly requires first complementary components containing two or more interaction sites capable of establishing multiple connections, and second the reversibility of the connecting events in order to allow the full exploration of the energy hypersurface of the system.[2]
Several synthetic strategies may be devised for the construction of macrobicyclic systems. The more direct one is the tripod-tripod coupling, a molecular self-assembly process that requires the formation of three bonds in a single step.[3] A major drawback of tripod-tripod coupling is the suffering of extensive side reactions which contribute to minimize the yield of the expected bicyclic product. Only in limited cases[4] such processes have been carried out in synthetically useful yields, provided a fine tuning of reagents, reactions and conditions could be achieved.
The imination reaction of tervalent phosphorus compounds with organic azides was discovered by H. Staudinger in 1919.[5] This reaction, known as the Staudinger reaction, is a two-step process that involves an initial nucleophilic addition of the P(III) centre of the phosphorus compound R13P to the terminal nitrogen of the organic azide R2N3, followed by dinitrogen elimination from the intermediate R13PN3R2 giving a l5-phosphazene R13P=NR2 (Scheme 1).

Only in a few instances the intermediate R13PN3R2, named by the same autor as phosphazide, has been isolated.[5b,6] Normally, this occur when phosphorus(III) compounds bearing substituents that increase the electron density at the phosphorus atom, or azides bearing electron-withdrawing substituents at the Na of the azido group were used. In that way, the intermediate phosphazide is stabilised (see Scheme 1: zwitterionic character of the PN3 framework in the dipolar canonical form). Also some phosphazides have been isolated starting from phosphanes or azides with very bulky substituents that prevent the formation of the l5-phosphazene, or when an intramolecular hydrogen bond may be formed in the phosphazide (Scheme 2). However, in any of such isolated phosphazides the PN3 fragment forms part of a ring.
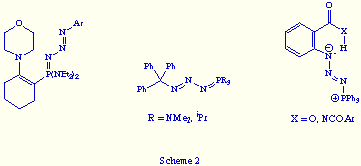
On the other hand, it is important to point up that the X-ray structural data of most of the reported phosphazides revealed the above mentioned zwitterionic character of the PN3 fragment, and the almost exclusive E configuration of the central N=N bond (see Scheme 1: dipolar canonical form).
Recently we reported[7] the preparation of the first two examples of macrobicyclic tris(phosphazides) by coupling of two tripodal subunits, a tris(o-azidobenzyl)amine and 1,1,1-tris[(diphenylphosphino)methyl]ethane (triphos), in the usual conditions of the Staudinger reaction (Scheme 3).
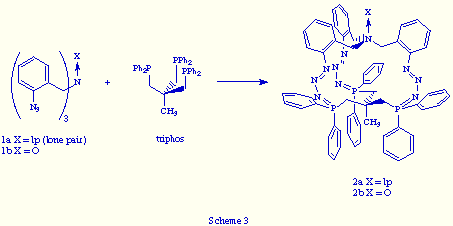
The structure of the N-oxide 2 (X= O) was unequivocally determined by X-ray analysis, the most important features of its structure being the following:
![]() The oxygen and the CH3 group at the bridgehead atoms are directed outward from
the bicyclic cavity (Figure 1)
The oxygen and the CH3 group at the bridgehead atoms are directed outward from
the bicyclic cavity (Figure 1)
![]() The three zwitterionic phosphazide units have Z configuration around
the central N=N bond (Figure 1)
The three zwitterionic phosphazide units have Z configuration around
the central N=N bond (Figure 1)
![]() This compound is chiral due to its propeller-like shape (Figure 1).
This compound is chiral due to its propeller-like shape (Figure 1).
| Figure 1 |
In this poster we disclose our recent results derived from the study of tripod-tripod coupling processes between other tris(azides) and tris(phosphanes) which have given rise to the design and synthesis of additional examples of macrobicyclic tris(phosphazides).
![]() Results and Discussion
Results and Discussion
![]() In attempts to explore the structural and electronic requirements of the
essential starting materials for an effective self-assembly, we have tested the
tripod-tripod coupling of other accesible tris(azides) and tris(phosphanes).
The first attempt was carried out reacting 1,1,1-tris(azidomethyl)ethane with
1,1,1-tris[(diphenylphosphino)methyl]methane (triphos) to yield a white solid.
When analyzed spectroscopically it revealed as an oligomerization product
(Scheme 4).
In attempts to explore the structural and electronic requirements of the
essential starting materials for an effective self-assembly, we have tested the
tripod-tripod coupling of other accesible tris(azides) and tris(phosphanes).
The first attempt was carried out reacting 1,1,1-tris(azidomethyl)ethane with
1,1,1-tris[(diphenylphosphino)methyl]methane (triphos) to yield a white solid.
When analyzed spectroscopically it revealed as an oligomerization product
(Scheme 4).
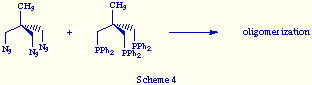
This result is indicative that the structural similarity between both reactants is not enough guarantee of an efficient self-assembly.
An analogous result was obtained when we reacted tris(o-azidobenzyl)amine 1a, the aromatic tris(azide) precursor of the phosphazide 2a, with tris(diphenylphosphino)methane (Scheme 5).
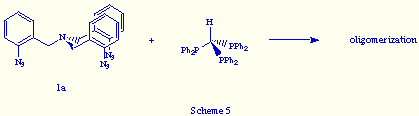
This result revealed that the use of aromatic tris(azides) do not assure an effective self-assembly.
These unsuccessful results showed that the occurrence of the self-assembly requires the convergence of several factors not yet well understood. The successful preparation of 2 was in fact a case of serendipity.
![]() To confirm the perfect complementarity of the reactants in the self-assembly
process leading to 2a in front of other side reactions, e. g.
oligomerization, we have carried out different crossed experiments. First, we
have tested the tripod-tripod coupling of tris(o-azidobenzyl)amine
1a with a mixture of triphos and tris(diphenylphosphino)methane, giving
as only product the phosphazide 2a (49% yield) (Scheme 6).
To confirm the perfect complementarity of the reactants in the self-assembly
process leading to 2a in front of other side reactions, e. g.
oligomerization, we have carried out different crossed experiments. First, we
have tested the tripod-tripod coupling of tris(o-azidobenzyl)amine
1a with a mixture of triphos and tris(diphenylphosphino)methane, giving
as only product the phosphazide 2a (49% yield) (Scheme 6).
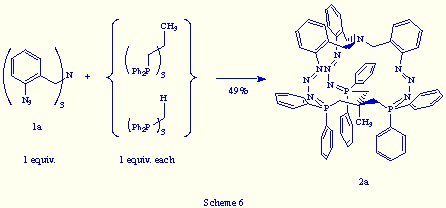
In the same way, the treatment of an equimolecular mixture of tris(o-azidobenzyl)amine 1a and 1,1,1-tris(azidomethyl)ethane with triphos, led to the phosphazide 2a in 75% yield (Scheme 7).
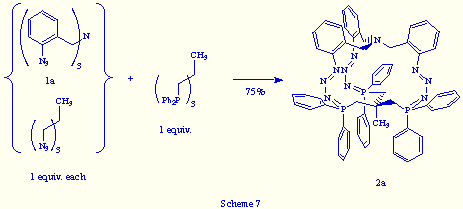
From these last two experiments, we conclude that a perfect recognition between both reactants, tris(o-azidobenzyl)amine 1a and triphos, is achieved at the molecular level and that the self-assembly mechanism that gives rise to the phosphazide 2a looks in fact like a "strict self-assembly"[8] of a multi-bridged cage molecule.
![]() In order to achieve other successful coupling reactions a more careful design
of the reaction partners have to be accomplished.
In order to achieve other successful coupling reactions a more careful design
of the reaction partners have to be accomplished.
1. The easiest way: tris(o-azidobenzyl)amines 1 and a tris(phosphane) strictly similar to triphos, e.g. tris[(diphenylphosphino)methyl]methane [HC(CH2PPh2)3].[9] The reactions gave rise to the corresponding macrobicyclic tris(phosphazides) 3 (X= lp, O) in good yields (Scheme 8).
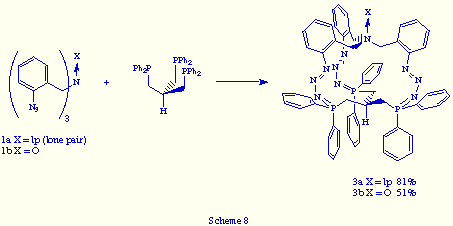
Their analytical and spectra data are essentially analogous to those obtained for tris(phosphazides) 2a,b. Their 31P{1H} NMR spectra showed a singlet at d= 3.62 ppm for compound 3a (X = lp) and d= 2.06 for 3b (X = O). Their 1H NMR spectra showed, in both tris(phosphazides) 3, the CH2N and CH2P hydrogen atoms as diastereotopics. The proton at the brigehead carbon atom appeared as a broad multiplet at d = 1.7 ppm. Thereby we propose that compounds 3 possess tridimensional structures similar to those of 2a,b, that is of propeller-like shape, with the lone pair or oxygen atom at the bridgehead nitrogen directed outward from the cavity, as well as the hydrogen atom at the bridgehead carbon atom, and the three phosphazide units of Z configuration around the central N=N bond.
2. Something more elaborated: triphos and other tris(azides) structurally
related to 1 which could give rise to the same relative positioning of
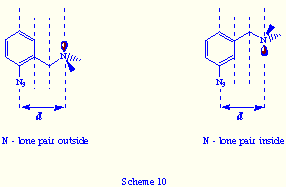
their three azido groups, e.g.: tris(m-azidobenzyl)amines 4![]() . The
mayor difference, respect to the ortho-substituted tris(azides), is that
in the self-assembly product (if formed) from 4 and triphos, the lone
pair or the oxygen atom will be positioned inward the bicyclic cavity (Scheme
10).
. The
mayor difference, respect to the ortho-substituted tris(azides), is that
in the self-assembly product (if formed) from 4 and triphos, the lone
pair or the oxygen atom will be positioned inward the bicyclic cavity (Scheme
10).
The reaction of tris(m-azidobenzyl)amine 4a with triphos was achieved by simultaneous addition of two ethereal solutions of both reagents to a flask containing diethyl ether, leading to a pale yellow solid. The structure determination of the isolated product was accomplished according to its spectral and analytical data, which were in good agreement with the tris(phosphazide) structure 5, and closely similar to those of 2a (Scheme 11).
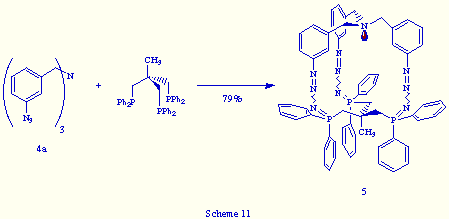
The only significant difference between 2a and 5 was the broad resonance around d= 4 ppm observed in the 31P{1H} NMR spectrum of 5 in CDCl3 at room temperature, contrary to the sharp singlet near d= -1 ppm shown by compound 2a in similar recording conditions. The broad signal may be indicative of the fluxional behaviour of 5 in CDCl3 solution at that temperature, which must be consequence of the lengthening by one carbon atom in each arm of the bicyclic skeleton of 5 in relation to 2a. In conclusion, the new tris(phosphazide) 5 is less rigid than 2a.
The tris(phosphazide) 5 is stable in CDCl3 solution as shown by 1H NMR and 31P{1H} NMR, and remained unchanged at room temperature after several weeks, contrary to its analogous ortho 2a which decompose totally in less than one day.
This stability of 5 in solution allowed us to obtain suitable crystal for X-ray analysis, by slow diffusion of diethyl ether into a dichloromethane solution of the crude reaction product. The study of the crystallographic data revealed the main features of its structure:
![]() The CH3 group of the triphos fragment is located outside of the bicyclic
cavity.
The CH3 group of the triphos fragment is located outside of the bicyclic
cavity.
![]() The lone pair at the brigehead nitrogen is oriented inside of the bicyclic
cavity.
The lone pair at the brigehead nitrogen is oriented inside of the bicyclic
cavity.
![]() Two of the phosphazide fragments have Z configuration with respect to
the central N=N (fragment containing P1 and P2), and the third (containing P3)
is of E configuration (see Figure 2)
Two of the phosphazide fragments have Z configuration with respect to
the central N=N (fragment containing P1 and P2), and the third (containing P3)
is of E configuration (see Figure 2)
![]() This compound is chiral, and its propeller-like shape is clearly apparent
when the molecule is viewed along the C5-C1-N1 vector (Figure 2).
This compound is chiral, and its propeller-like shape is clearly apparent
when the molecule is viewed along the C5-C1-N1 vector (Figure 2).
| Figure 2 |
While, as it has been just showed, the reaction of 4a and triphos give the phosphazide 5, the behaviour of the N-oxide 4b derived from tris(m-azidobenzyl)amine is quite different. Its reaction with triphos did not give any self-assembly product but yielded a solid that is probably an oligomerization product, according to its spectral data (Scheme 12).
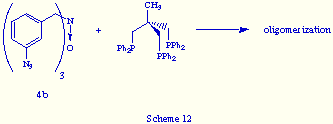
We envisage that the unsuccessful coupling of 4b and triphos may be a consequence either:
a) of a decrease of the population of the optimal reactive conformer for the coupling process, following the quaternization of the nitrogen atom of 4a.
or/and
b) due to the fast decomposition of the resultant macrobicyclic tris(phosphazide). This unstability would be originated by the inside location of the oxygen atom in the bicyclic cavity.
At the present time, we are designing and synthesizing new tripodal tris(azides) and tris(phosphanes) capable to give rise to self-assembly products with the aim of obtaining new clues for the rational statement of the structural and electronic factors that control these processes.
![]() Acknowledgements
Acknowledgements
Thanks are given to Dirección General de Investigación Científica y Técnica for financial support (project number PB95-1019).