Synthesis of 1,2-Dialkyl-perhydro-quinolin-6-ones: A One Pot Birch Reduction as Key Step
Institute of Organic Chemistry
Vienna University of Technology, Getreidemarkt 9, A-1060 Vienna, Austria (EU)
Introduction
The Birch reduction is a well established method for the conversion of
aromatic cores to the corresponding dihydro compounds. Due to the low stability
of the resulting enamine moiety this reaction causes problems when applied
to some nitrogen containing heterocycles. However, in many cases these
enamines serve as intermediates for a complete reduction of the aromatic
core en route to the corresponding perhydro system. Procedures reported
in the recent literature suffer from several disadvantages ranging from
low yields (1), only partial reduction of the heterocyclic system (2),
complex product mixtures (3), high pressure techniques (4), to rather complex
and time consuming multi step approaches (5). In the present publication
we would like to present our first results in applying the one-pot Birch
reaction developed for the synthesis of 4-dimethylaminocyclohexanone (6)
to the quinoline system en route to the title compounds.
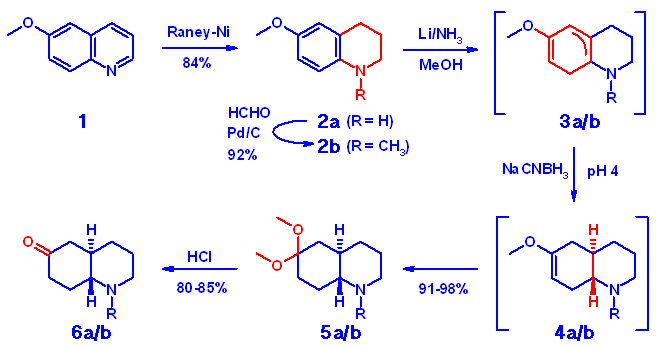
1-Substituted Quinoline-Precursors
Precursors 2a/b were accessible in high yield by Raney-Ni reduction
of quinoline 1 followed by reductive methylation and exhibited a
comparable behavior during the reduction process to the model compound
4-dimethylaminocyclohexanone. For both N-substituted and unsubstituted
compounds 2a and 2b the Birch protocol including a subsequent
NaCNBH3 reduction step in a one-pot procedure gave an excellent
to quantitative yield of reduced ketals 5a and 5b, respectively,
representing a significant improvement to all methods known to us. Conversion
to ketones 6a/b was carried out according to standard procedures.
Absolute selectivity of the stereochemistry at the annelation site in favor
of the trans product was observed and no traces of any cis-isomer
were detected.
Initial reduction of the precursors 2a/b with lithium gives the
intermediates 3a/b, which are converted to the enolethers 4a/b
by treatment with NaCNBH3 at pH 4. All intermediates
were detected by NMR-spectroscopy.
1,2-Disubstituted Quinoline-Precursors
For the synthesis of appropriate 1,2-disubstituted precursors we extended
a procedure for the Ziegler-type addition of organolithium compounds to
quinolines reported recently (7). The intermediate 7 of the initial
attack in two position was trapped by quenching with MeI to give substitution
at the nitrogen in a one pot reaction. Crude 8 was further reduced
to give tetrahydro-compound 9 as substrate for the Birch/NaCNBH4
process.
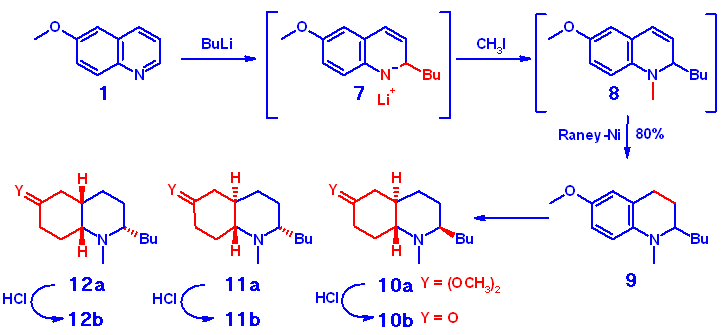
In contrast to the unsubstituted aromatic heterocycle the formation
of considerable amounts of the cis-fused system 12a was observed.
We attribute this effect to a steric interaction of the sidechain with
the hydrid reagent. Obviously the main attack vector of the reducing agent
is blocked to some extent by the carbon chain. Therefore the energy difference
between the two possible sides for the approach to the intermediate immonium
structure is decereased, resulting in a mixture of cis- and trans-isomers.
It is noteworthy that the major isomer formed is the trans-axial
compound 10a. It seems likely that the protonation step during the
NaCNBH3 reduction is responsible for the
different diastereochemistry at C-2. Introduction of a proton at C-4a results
in a significant change of conformation according to calculations performed
on semiempirical level, directing the side chain either into the axial
or equatorial position. However, the energy difference is small,
and the observed mixture is the result: only one 2-axial isomer
ö trans-10a ö was isolated as the main product in 41% yield,
while both 2-equatorial compounds 11a (18%) and 12a
(10%) were observed. Calculations based on refined models are underway
in order to provide more insight into the formation of the 3 diasteromers.
Further studies to investigate the influence of chain length as well
as introduction of branched substituents on the stereochemistry of the
Birch/NaCHBH3 sequence are in progress within our
lab.
Experimental
Raney-Ni Reduction
A solution of the quinoline 1 in MeOH/KOH was treated with Raney-Ni and
gently refluxed until complete conversion. A general workup initiated by
filtration through Celite gave the pure product after Kugelrohr distillation.
One Pot Birch/NaCNBH4 Reduction
A 30% solution of 2a/b (1 equiv.) in dry THF and dry MeOH (10 equiv.) was
added to dry liquid ammonia condensed directly into the reaction vessel
to form an approx. 10% reaction solution. Lithium chips (10 equiv.) were
added slowly, maintaining the temperature at -35 ± 5°C. After
the initially vigorous reaction had ceased the mixture was refluxed until
the blue color disappeared. Ammonia and the organic solvents were evaporated
by a stream of nitrogen at approx. 50°C and the residue was treated
with dry THF and MeOH. The resulting solution was cooled to 5°C and
brought to pH 4 by addition of methanolic HCl using bromocresol green as
indicator. During the addition of NaCNBH3 pH 4 was maintained by treatment
with methanolic HCl. When the pH showed no further change solid NaHCO3
and some NaOH were added and the solvents evaporated. The crude product
obtained by a general work-up was refluxed overnight in a mixture of MeOH
and methanolic HCl. Isolation of pure 5a/b was performed by Kugelrohr distillation.
1,2-Ziegler-type Substitution and Reduction of
the Heterocyclic Core
A solution of n-BuLi (1 eq.) in dry THF was treated with 1 (1 eq.)
in dry THF at ö20°C, slowly brought to rt and stirred for one hour.
Treatment with CH3I (2.5 eq) at 0°C, stirring for another
hour at rt, followed by a general workup gave 8 without further purification
and ready for the Raney-Ni reduction.
Acknowledgement
 We
would like to thank NOVARTIS Crop Protection Basel for generously funding
this project.
We
would like to thank NOVARTIS Crop Protection Basel for generously funding
this project.

References
-
Hönel, M.; Vierhapper, F.W. J. Chem. Soc, Perkin Trans. I 1980,
1933.
-
O'Brien, S.; Smith, C.C. J. Chem. Soc. 1960, 4609; Remers,
W.A.; Gibs, G.J.; Pidacks, C.; Weiss, M.J. J. Am. Chem. Soc. 1967,
89, 5513; Remers, W.A.; Gibs, G.J.; Pidacks, C.; Weiss, M.J. J.
Org. Chem. 1971, 36, 279.
-
Sagatova, Y.K.; Karimov, M.K.; Yunusov, T.K.; Safaev, A.S. Deposited
Doc. 1984, 907; Chem. Abstr. 102: 149083m.
-
Mehta, P.; Kumar, Y.; Saxena, A.K.; Gulati, A.K.; Singh, H.K; Anand, N.
Ind. J. Chem. 1991, 30B, 213.
-
Schaus, J.M.; Huser, D.L., Titus, R.D. Synth. Commun. 1990,
20, 3553; Schaus, J.M. 1985, US 4540787 - Chem. Abstr. 109:
270253w; Schaus, J.M. 1984, JP 59222477 - Chem. Abstr. 102:
166627j; Schaus, J.M. 1984, EP 127708 - Chem. Abstr. 102:
166626h.
-
Stanetty, P.; Kasemann, O.; Schmid, M. J. Chem. Research(S) 1995,
341.
-
Goldstein, S.W.; Dambek, P.J. Synthesis 1989, 221.