| [Molecules: 2] [Related articles/posters: 097 083 112 009 052 ] |

Introduction
(-)-Balanol 1 is a potent inhibitor of protein kinase C, a family of
phospholipid dependent serine/threonine protein kinases which play an important
role in cell growth, signal transduction and differentiation. The activated
enzyme has been implicated in many diseases, such as cancer, inflammation and
HIV infection. (-)-Balanol 1 was initially isolated by workers at
Sphinx Pharmaceuticals from the fungus Verticillium balanoides, [1] and recently from species of Fusarium [2] by workers at Nippon Roche. The remarkable inhibitory properties toward protein kinase C displayed by 1 have stimulated recent synthetic efforts culminating in several total syntheses of 1 and all synthetic methods reported involve the connection of two distinct structural domains, chiral hexahydroazepine-containing
fragment [3-8] and benzophenone fragment. [3-5]
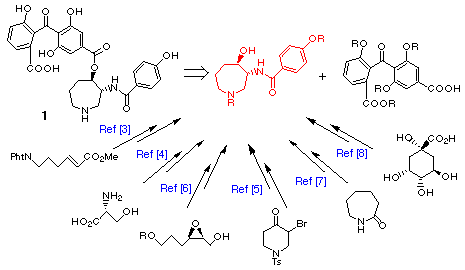
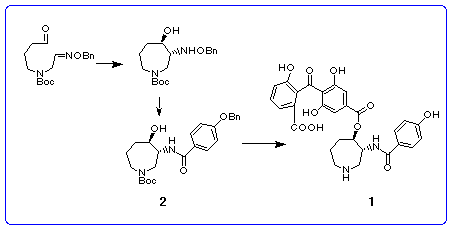
In this report we wish to describe a novel and concise synthesis of both enantiomers of the chiral hexahydroazepine-containing fragment via the route involving the radical cyclization of the aldehyde connected with oxime ether followed by optical resolution of the racemic product.
Radical cyclization
Free radical cyclization has been an important tool for the construction of various types of cyclic compounds including biologically active natural products and medicinals. [9] There have been several examples involving radical cyclizations of olefins and alkynes having aldehydes or ketones in the presence of tributyltin hydride. [10] In continuation of our study on sulfanyl radical addition-cyclization of dienes, [11] we developed the first example of tributyltin hydride-induced cyclization of oxime ethers [12] which are intramolecularly connected to aldehydes or ketones. [13] The newly found radical cyclization provides a synthetically useful
method for the construction of cyclic amino alcohols widely found in biologically active natural products such as amino sugars, aminocyclitols, [14] and sphingolipids.
A solution containing tributyltin hydride (2 equiv.) and AIBN (0.2 equiv.) in benzene was added dropwise to a solution of the oxime ether 3 (1 equiv.) in boiling benzene. The solution was then refluxed for a further several hours and the solvent was removed in vacuo. The resulting residue was purified by medium pressure column chromatography to give the cyclized products 4 and 5. Several examples of the cyclization are shown in Table 1. The radical cyclizations proceeded very smoothly to give five- to seven- membered cyclic amino alcohols in good yields of which trans-isomers 5 were obtained as a major product. The newly found radical cyclization was extended to the oxime ether having terminal ketone 3e which gave effectively two cyclic amino alcohols 4e and 5e. Radical cyclizations in the formations of seven-membered products 4d and 5d and tertiary alcohols 4e and 5e proceeded slowly to recover the starting compounds in 35-46% yields, but addition of 1 equiv. of AIBN to the reaction mixture gave the improved yields shown in Table 1. The stereostructures of products 4 and 5 were firmly established by the chemical conversions involving the formation of the corresponding cyclic ketals in the cis-isomers 4.
| Substrate | R | m | n | Reaction time | Ratio 4 : 5 | Combined yield |
| 3a | H | 1 | 1 | 5 h | 30 : 70 | 54% |
| 3b | H | 2 | 1 | 6 h | 39 : 61 | 62% |
| 3c | H | 1 | 2 | 8 h | 20 : 80 | 70% |
| 3d | H | 3 | 1 | 9 h | 32 : 68 | 44% (70%)a |
| 3e | Me | 1 | 1 | 9 h | 21 : 79 | 52% (65%)a |
|
||||||
Thus, a general method for the intramolecular cyclization of oxime ethers
tethered to the carbonyl groups has been developed. The application of this
method to the synthesis of the hexahydroazepine 2, key intermediate
for the synthesis of (-)-balanol 1 is described in the next section.
Synthesis of (±)-hexahydroazepine
In order to apply the radical cyclization described above to the synthesis of
the known hexahydroazepine 2, [3] we chose the radical precursor
8 which has the Boc group on nitrogen and the benzyloxyimino
group.
4-Aminobutanol 6 was alkylated with -chloroacetaldoxime benzyl ether, [15] readily prepared from the corresponding aldehyde and
benzyloxyamine, to give the secondary amine which was acylated with
di-tert-butyl carbonate under Schotten-Baumann conditions to afford the
hydroxy oxime ether 7 in 67% yield from 6. Mild
oxidation of the alcohol 7 with chromium(VI) oxide-pyridine gave the
unstable aldehyde 8 in 77% yield. The compounds 7 and 8 were the mixtures of E- and Z-isomers (3:2) in the geometry of the oxime ether group, and used in the following
radical cyclization without separation. Stannyl radical
cyclization of the aldehyde 8 by treatment with
tributyltin hydride (2 equiv.) in the presence of AIBN (1 equiv.) proceeded
smoothly to give a 1:2.5 mixture of two cyclized products
9 and 10 in 58% combined yield
which was readily separated by medium pressure column chromatography.
-chloroacetaldoxime benzyl ether, [15] readily prepared from the corresponding aldehyde and
benzyloxyamine, to give the secondary amine which was acylated with
di-tert-butyl carbonate under Schotten-Baumann conditions to afford the
hydroxy oxime ether 7 in 67% yield from 6. Mild
oxidation of the alcohol 7 with chromium(VI) oxide-pyridine gave the
unstable aldehyde 8 in 77% yield. The compounds 7 and 8 were the mixtures of E- and Z-isomers (3:2) in the geometry of the oxime ether group, and used in the following
radical cyclization without separation. Stannyl radical
cyclization of the aldehyde 8 by treatment with
tributyltin hydride (2 equiv.) in the presence of AIBN (1 equiv.) proceeded
smoothly to give a 1:2.5 mixture of two cyclized products
9 and 10 in 58% combined yield
which was readily separated by medium pressure column chromatography.
We then examined the intramolecular reductive coupling of the aldehyde with oxime ether in 8, in the presence of samarium diiodide. This one-electron reducing agent has been previously used for the intermolecular reductive coupling of aldehydes and O-benzylformaldoxime. [16] Upon treatment with samarium diiodide in THF containing HMPA and ethylene glycol, the aldehyde 8 (a mixture of E : Z=3:2) gave almost the same result, affording a 1:3 mixture of two products 9 and 10 (35% combined yield), as in the radical cyclization promoted by tributyltin hydride.
Hydrogenolysis of the benzyloxyamino group in trans-product 10 in the presence of platinum dioxide followed by N-acylation with p-(benzyloxy)benzoyl chloride afforded the desired azepine 2 in 58% yield from 10. Thus, we have succeeded in the six-step synthesis of the racemic key intermediate 2 [3] for the synthesis of balanol. [17]
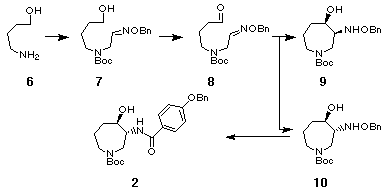
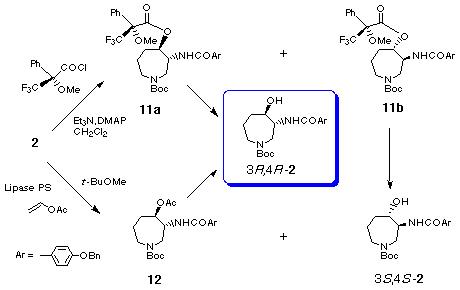
In order to explore a new method for the large scale preparation of enantiomerically pure hexahydroazepine 2, we next investigated the enzymatic esterification of the racemic alcohol 2 or enzymatic hydrolysis of the racemic acetate 12. Among the conditions investigated, the enzymatic esterification [19] of 2 provided (3R,4R)-acetate 12 with 99% ee (42%) and the recovered (3S,4S)-2 82% ee (49%) using Lipase PS (Wako, Lipase, immobilized, from Pseudomonas sp.) with vinyl acetate as an acylating in ButOMe. The attempted hydrolysis of the racemic acetate 12 by using Lipase PS was unsuccessful and the racemic acetate 12 was completely recovered. Thus we have now succeeded in the chiral synthesis of both enantiomers of the key intermediate 2 of which (3R,4R)-isomer 2 was prepared in large scale via the route involving enzymatic acetylation.
Our chiral synthesis of the key intermediate 2 and the enantiomer would open a novel asymmetric synthetic route for (-)- and (+)-balanols.
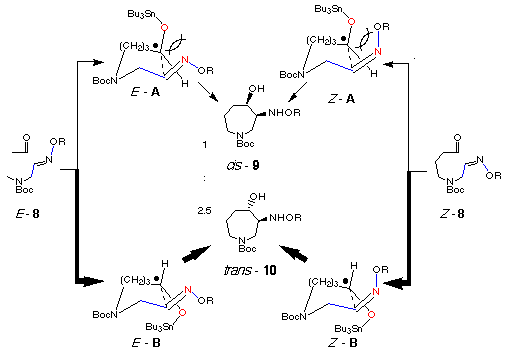
As mentioned above, we employed a mixture of E- and Z-oxime ethers as a substrate for the radical cyclization. In order to clarify the relationship between the stereoselectivity and the geometry of the oxime ethers of the starting substrate 8, we separated both isomers by medium pressure column chromatography and undertook the radical cyclization. The results obtained are collected in Table 2 which shows almost the same stereochemical outcome irrespective of the geometry of the starting E- and Z-oxime ethers 8. Recently a similar result has been reported in the reductive coupling reaction of carbonyl group with oxime ethers using samarium diiodide. [21]
| Substrate | Ratio
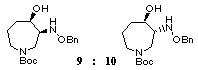 | Combined Yield |
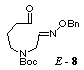 | 1 : 2.8 | 51% |
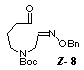 | 1 : 2.7 | 43% |
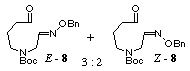 | 1 : 2.5 | 50% |
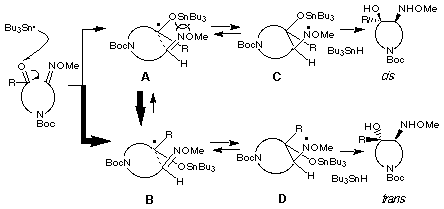
Considering our result of trans-selectivity of the products irrespective of geometry of the starting oxime ether and the well-known no thermal isomerization of the oxime ether, we propose again the reaction pathway of the radical cyclization as shown in the following Scheme. Two groups of transition states E- and Z-A and E- and Z-B are proposed for giving cis- and trans-products, respectively. In both cases, transition states E- and Z-A would be more unstable than transition states B due to the electronic repulsion between stannyloxy and nitrogen of oxime ether. There would be no influence of geometry of oxime ether group in all four transition states because alkoxy group of the oxime ethers is not close to the stannyloxy group. Therefore, trans-isomer 10 would be formed as a major product via the transition states E- and Z-B.
 : 7.45-7.25 [28/5H, m, ArH and HC=N (E)], 6.70 [2/5H, br
t, J=4 Hz, HC=N (Z)], 5.11 [4/5H, s, CH2Ph (Z)],
5.07 [6/5H, s, CH2Ph (E)], 4.15-4.00 [4/5H, m, CH2CH=N
(Z)], 4.00-3.80 [6/5H, m, CH2CH=N (E)], 3.65-3.55 (2H, m,
CH2OH), 3.30-3.10 (2H, m, CH2CH2NBoc), 1.60-1.40 (4H, m,
CH2CH2CH2NBoc), 1.44 (9H, s, Me x 3). MS Calc for C18H28N2O4
(M+); 336.2047. Found: 336.2049.
: 7.45-7.25 [28/5H, m, ArH and HC=N (E)], 6.70 [2/5H, br
t, J=4 Hz, HC=N (Z)], 5.11 [4/5H, s, CH2Ph (Z)],
5.07 [6/5H, s, CH2Ph (E)], 4.15-4.00 [4/5H, m, CH2CH=N
(Z)], 4.00-3.80 [6/5H, m, CH2CH=N (E)], 3.65-3.55 (2H, m,
CH2OH), 3.30-3.10 (2H, m, CH2CH2NBoc), 1.60-1.40 (4H, m,
CH2CH2CH2NBoc), 1.44 (9H, s, Me x 3). MS Calc for C18H28N2O4
(M+); 336.2047. Found: 336.2049.
8: 1H NMR
(200 MHz, CDCl3) : 9.74 [2/5H, t, J=1 Hz, CHO (Z)], 9.71
[3/5H, t, J=1 Hz, CHO (E)], 7.41-7.20 (5H, m, ArH), 7.40 [3/5H, br
t, J=4 Hz, HC=N (E)], 6.69 [2/5H, br t, J=4 Hz, HC=N
(Z)], 5.11 [4/5H, s, CH2Ph (Z)], 5.06 [6/5H, s,
CH2Ph (E)], 4.15-4.00 (4/5H, m, CH2CH=N (Z)),
3.98-3.82 (6/5H, m, CH2CH=N (E)), 3.30-3.12 (2H, m,
CH2CH2NBoc), 2.50-2.30 (2H, m, CH2CHO), 1.90-1.70 (2H, m,
CH2CH2NBoc), 1.44 (9H, s, Me x 3). MS Calc for C18H26N2O4
(M+); 334.1891. Found: 334.1889.
9: 1H NMR (200 MHz,
CDCl3) : 7.42-7.28 (5H, m, ArH), 4.70 (2H, s, CH2Ph), 4.05 (1H,
m, 4-H), 3.66-3.07 (5H, m, 2-H2, 3-H, 7-H2), 2.08-1.55 (4H, m, 5-H2, 6-H2),
1.44 (9H, s, Me x 3). MS Calcd for C18H28N2O4 (M+); 336.2047. Found:
336.2036.
10:
1H NMR (200 MHz, CDCl3) : 7.42-7.25 (5H, m, ArH), 4.70 and
4.68 (2H, ABq, J=14 Hz, CH2Ph), 3.71-2.83 (6H, m, 2-H2, 3-H, 4-H,
7-H2), 2.00-1.40 (4H, m, 5-H2, 6-H2), 1.44 (9H, s, Me x 3). MS Calc for
C18H28N2O4 (M+); 336.2047. Found: 336.2032.
(3R,4R)-2: Colourless needles (MeOH), mp
136-138 oC. []D-2.9 (c =1.30, MeOH). [Lit., [18] mp
115-117 oC. []D-8.9 (c =1.98, MeOH)].
(3S,4S)-2: colourless needles (MeOH), mp
136-138 oC. []D +2.5 (c =1.26, MeOH).
References
[1] Kulanthaivel, P.; Hallock, Y. F.; Boros, C.;
Hamilton, S. M.; Janzen, W. P.; Ballas, L. M.; Loomis, C. R.; Jiang, J. B.;
Katz, B.; Steiner, J. R.; Clardy, J. J. Am. Chem. Soc. 1993,
115, 6452.
[2] Ohshima, S.; Yanagisawa, M.; Katoh, A.; Fujii, T.; Sano, T.; Matsukuma, S.; Furumai, T.; Fujiu, M.; Watanabe, K.; Yokose, K.; Arisawa, M.; Okuda, T. J. Antibiotics 1994, 47, 639.
[3] Lampe, J. W.; Hughes, P. F.; Biggers, C. K.; Smith, S. H.; Hu, H. J. Org. Chem. 1994, 59, 5147.
[4] Nicolaou, K. C.; Bunnage, M. E.; Koide, K. J. Am. Chem. Soc. 1994, 116, 8402.
[5] Adams, C. P.; Fairway, S. M.; Hardy, C. J.; Hibbs, D. E.; Hursthouse, M. B.; Morley, A. D.; Sharp, B. W.; Vicker, N.; Warner, I. J. Chem. Soc., Perkin Trans. 1 1995, 2355.
[6] Tanner, D.; Almario, A.; Högberg, T. Tetrahedron 1995, 51, 6061.
[7] Hu, H.; Jagdmann Jr., G. E.; Hughes, P. F.; Nichols, J. B. Tetrahedron Lett. 1995, 36, 3659.
[8] Albertini, E.; Barco, A.; Benetti, S.; De Risi, C.; Pollini, G. P.; Zanirato, V. Synlett 1996, 29.
[9] "Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds," Giese, B. Pergamon Press, New York, 1986; Curran, D. P. Synthesis 1988, 417 and 489.
[10] Hays, D. S.; Fu, G. C. J. Am. Chem. Soc. 1995, 117, 7283 and references therein.
[11] Naito, T.; Honda, Y.; Miyata, O.; Ninomiya, I. J. Chem. Soc., Perkin Trans. 1 1995, 19.
[12] Corey, E. J.; Pyne, S. G. Tetrahedron Lett. 1983, 24, 2821; Hart, D. J.; Seely, F. L. J. Am. Chem. Soc. 1988, 110, 1631; Bartlett, P. A.; McLaren, K. L.; Ting, P. C. J. Am. Chem. Soc. 1988, 110, 1633; Parker, K. A.; Spero, D. M.; Epp, J. V. J. Org. Chem. 1988, 53, 4628; Enholm, E. J.; Burroff, J. A.; Jaramillo, L. M. Tetrahedron Lett. 1990, 31, 3727; Booth, S. E.; Jenkins, P. R.; Swain, C. J. J. Chem. Soc., Chem. Commun. 1991, 1248; Barton, D. H. R.; Dalko, P. I.; Greco, S. D. Tetrahedron Lett. 1991, 32, 4713; Hatem, J.; Henriet-Bernard, C.;Grimaldi, J.; Maurin, R. Tetrahedron Lett. 1992, 33, 1057; Marco-Contelles, J.; Destabel, C.; Gallego, P.; Chiara, J. L.; Bernabé, M. J. Org. Chem. 1996, 61, 1354 and references therein.
[13] Naito, T.; Tajiri, K.; Harimoto, T.; Ninomiya, I.; Kiguchi, T. Tetrahedron Lett. 1994, 35, 2205.
[14] Kiguchi, T.; Tajiri, K.; Ninomiya, I.; Naito, T.; Hiramatsu, H. Tetrahedron Lett., 1995, 36, 253.
[15] Stach L. J. U. S. Patent 1975, 3920772 [Chem. Abstr., 1976, 84, 89603d].
[16] Hanamoto, T.; Inanaga, J. Tetrahedron Lett. 1991, 32, 3555.
[17] Naito, T.; Torieda, M.; Tajiri, K.; Ninomiya, I.; Kiguchi, T. Chem. Pharm. Bull. 1996, 44, 624.
[18] Private communication from Dr. P. F. Hughes.
[19] Klibanov, A. M. Acc. Chem. Res. 1990, 23, 114.
[20] Shono, T.; Kise, N.; Fujimoto, T.; Yamanami, A.; Nomura, R. J. Org. Chem. 1994, 59, 1730.
[21] Chiara, J. L.; Marco-Contelles, J.; Khiar, N.; Gallego, P.; Destabel, C.; Bernabé, M. J. Org. Chem. 1995, 60, 6010.