C2-Symmetric and trans-chelating heterocyclic ligand,
4,4'-disubstituted (dibenzofuran-4,6-diyl)-2,2'-bioxazolines DBFOX. Applications to asymmetric Diels-Alder reactions
Shuji Kanemasa,
Yoji Oderaotoshi, Junji Tanaka and Hidetoshi Yamamoto
Institute of Advanced Material Study, Kyushu University, Kasugakoen,
Kasuga 816, Japan
Abstract
Several C2-symmetric bisoxazoline
chiral ligands in which two molecules of 4-chiral oxazolines are bound to
an appropriate spacer have been prepared. They have been designed to coordinate to a metal ion at
the trans-position so that the two chiral oxazolines can produce an effective
C2-symmetric space around the metal ion. The efficiency of the metal complexes
has been examined in asymmetric hydrosilylation and Diels-Alder reactions.
Introduction
the proposal of new methodologies by which highly effective
chiral induction can be accomplished are now strongly required in the field
of catalysed asymmetric synthesis. We now propose a new concept in the molecular
design of chiral ligands based on the structure of trans-chelating
ligands. Most known chiral ligands are cis-chelating. Their
metal complexes have a metal ion which is rather outside of the chiral sphere
made by the chiral ligand. This problem would be solved by introducing a
trans-chelating ligand. Some preliminary results are presented.
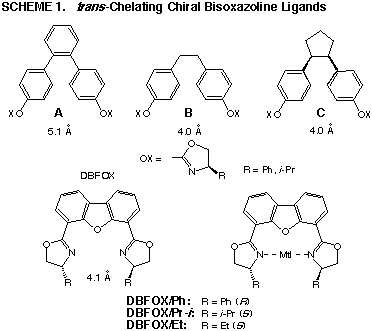
Molecular design
When two molecules of 4-chiral oxazoline rings
are bound to a spacer, a variety of chelating ligands are constructed. We
selected the four spacers shown in Scheme 1 in the present work. The nitrogen-nitrogen
distance is a little longer than 4 A so that metal ions can be tightly
captured between the nitrogens. Conformational rigidity of the ligand is
also important. On this basis, the DBFOX ligands which involve a dibenzofuran-4,6-diyl
spacer have been designed.
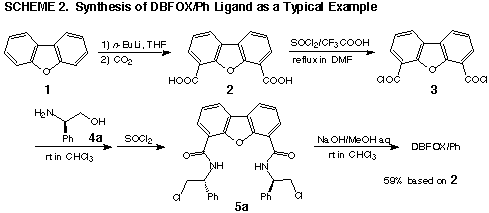
Ligand synthesis
Synthesis of the trans-chelating bisoxazoline
ligands starts from the corresponding dicarboxylic acids. Scheme 2 shows
the synthesis of (R,R)-dibenzofuran-4,6-diyl-bis(4-phenyloxazoline),
DBFOX/Ph as a typical example. Commercially available dibenzofuran 1 was
lithiated with an excess of butyllithium (3 equiv.) in THF and then treated
with carbon dioxide to give dicarboxylic acid 2. This acid 2 is very insoluble
in most organic solvents, therefore, a mixture of thionyl chloride and trifluoroacetic
acid in DMF was used for its transformation to acid chloride 3. Subsequent
reaction with an optically pure amino alcohol and then with thionyl chloride
gives chloride 5a. Cyclization of 5a with sodium hydroxide provides DBFOX/Ph
in 59% yield based on the dicarboxylic acid 2. Other ligands can be obtained
by similar procedures.
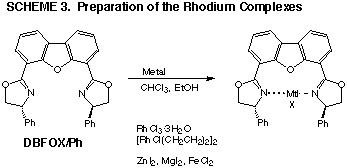
Complex formation
In our previous work, the RhIII and RhI
complexes were prepared by using ligands A-C. Although the complex formation
was confirmed by 1H NMR spectra in which the ring protons of the oxazoline
ring were deshielded, their thermodynamic stability was not high enough.
Even under heating in dichloromethane they were dissociated to their components
together with unidentified and highly insoluble materials. On the other
hand, DBFOX/Ph formed highly stable complexes with RhIII,
RhI, FeII,
Mg, and Zn ions whose formation was again confirmed by similar low field
shifts of the corresponding protons. They can be isolated, but their purification
by crystallization is so far unsuccessful.
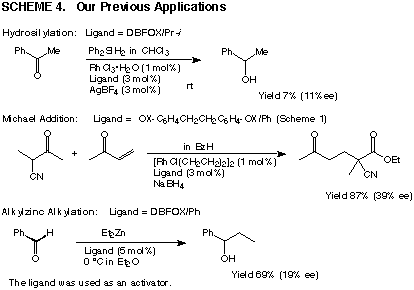
Catalysed reactions
Scheme 4 shows our previous results observed
in asymmetric reactions using trans-chelating ligands A-C
and DBFOXs. Although the RhIII/A-C complexes showed high catalytic activity
in hydrosilylation, racemic alcohols resulted. In contrast, the DBFOX
complexes are very inactive, indicating the high thermodynamic stability
of the complexes. The RhI/B complex accelerated the Michael addition between
2-methyl-3-oxobutanenitrile and 3-buten-2-one to afford a moderate chiral
induction, while the DBFOX complexes were again inactive. DBFOX ligands
accelerated the alkylation reactions between diethylzinc and benzaldehyde,
but the chiral induction was not effective.
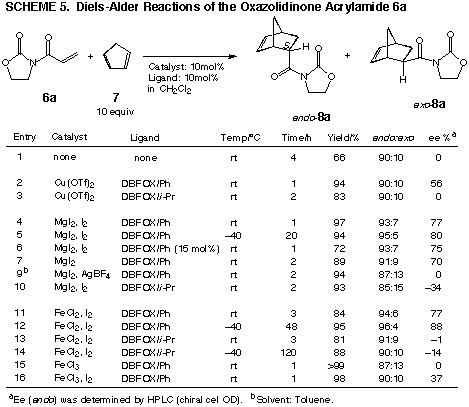
Diels-Alder reactions
Metal complex catalysts of DBFOXs were
prepared in situ with copper(II) triflate, magnesium iodide, and iron(III)
chloride. The resulting Lewis acid catalysts were utilized in the Diels-Alder
reactions of cyclopentadiene 7 with 3-acryloyl-2-oxazolidinone 6a as
shown in Scheme 5. The cation complexes of magnesium and iron ions were
effective catalysts in the reactions at p;40 °C. The shielding
substituent at the 4-position of the oxazoline has to be chosen properly,
a phenyl substituent being better than an isopropyl moiety. Although the magnesium
iodide complex showed a catalytic activity, iron(III) chloride complex did
not. Also important was the procedure for cation complexes. The method using
magnesium iodide + iodine provided an active catalyst, while AgBF4 was ineffective.
The major stereoisomer produced in the DBFOX/Ph-catalysed reaction was characterized
to be (4S)-bicyclo[2.2.1]hept-2-ene-4-carboxyamide endo-8a.
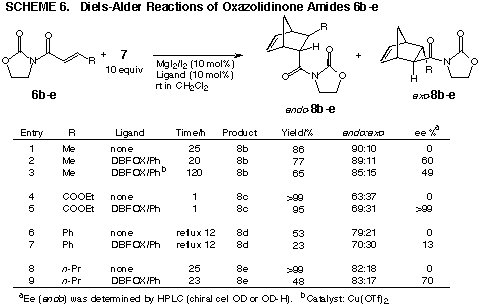
The cyclopentadiene Diels-Alder cycloadditions using other oxazolidinone
amides 6b-e in the presence of the DBFOX/Ph complex of MgI2/I2 were investigated
(Scheme 6). The reactive dienophile 6c showed an absolute chiral induction
even at room temperature, while the sluggish substrate 6d gave 8d in low
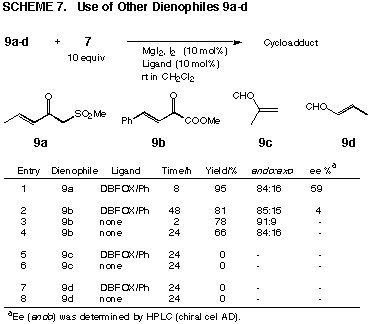
optical yield. Compared with the oxazolidinone amides 6a-e, other chelating
dienophiles 9a,b and reactive enals 9c,d resulted in unsatisfactory optical
yields (Scheme 7). Monodentate enals 9c,d especially could not be accelerated.
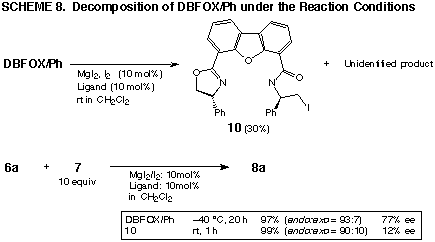
Stability of ligands
It was found that DBFOX ligands were gradually
decomposed under the reaction conditions using the magnesium complexes at
room temperature. One of the decomposition product was ring-opened 10 (Scheme
8). Although this compound 10 could act as a catalyst in the Diels-Alder reaction,
its chiral induction was much less effective than that of DBFOX/Ph. This
indicates that the efficiency of chiral induction should be lowered in the
latter stage of the reaction, especially if a longer reaction time is required.
Reaction mechanism
Selection of enantiofaces in the above Diels-Alder
reactions is well understood when the CHEM3D molecular models for the catalyst/dienophile
complexes are utilized. For the complex between the magnesium complex of
DBFOX/Ph and 3-acryloyl-2-oxazolidinone, two diastereomeric coordinations
of the dienophile are possible. The enantiofaces of the acryloyl moiety
are both hindered in the upper complex, while one of the enantiofaces is
freely open in the bottom complex. Accordingly, the reaction proceeds through
the bottom complex. Dependence of the optical yields observed upon the structures
of dienophiles is not so far clear. Any suggestions and comments concerning
this point are appreciated.