Summary: We propose a novel class of spiroaromatic
ring systems characterised by having a common phosphorus atom
in which each ring can independently exhibit either Möbius
or Hückel p-electron
aromaticity.
Conventionally constituted aromatic rings can fuse by
sharing a mutual bond between two sp2 hybridised
carbon or hetero atoms; (Hückel) ring aromaticity is
achieved by parallel overlap of the p-orbitals, forming p molecular orbitals with 4n+2 electron
occupancy. Although the normal maximum coordination for the
aromatic ring atoms is three, Wang and Schleyer1
have recently shown that four-coordinate phosphorins (1)
with e.g. X=F,Cl,OR, can also exhibit Hückel-like
p-aromaticity of the ring.
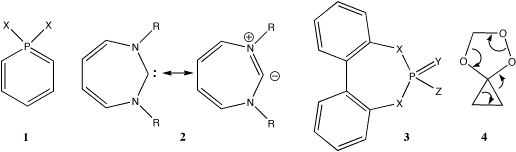
An alternative to parallel overlap of the p-orbitals is a 180o phase shift in the pp-pp overlap, (ideally) delocalised over the entire ring, to form a so-called Möbius system. This category of aromatic ring, first proposed by Heilbronner in 1964,2 has been much studied recently3-7. For example, the formal 8-p 4n electron ylid 2, adopts a C2 symmetric distortion as a stabilising mode to the otherwise planar 4n anti-aromatic Hückel form. Compounds derived by replacing the C-/carbene atom in 2 with a tri or tetracoordinate phosphorus atom as in 3 are known for both X=O8 and X=N9 where Y,Z can be either heteroatoms or be part of e.g. a cyclophosphazene ring. The latter example comprises what we term a spiroaromatic ring system, in which two aromatic rings are fused by sharing a single atom rather than by a mutual bond. The term spiroaromatic has been previously used10 to describe two independent aromatic ring systems joined by an sp3 carbon atom, which is itself not part of any aromatic ring; we propose here a definition which differs in that the e.g. P(V) spiro atom can itself participate in the aromatic conjugation.
Several of these systems illustrated by 3 exhibit significant chiral catalytic ability as e.g. metal ligands8h for enantionselective trapping of arene oxides. The chirality derives from C2-like symmetry of the 7-ring, but this is normally attributed to the steric effects of the two benzo groups, rather than deriving from aromatising stabilisation. Spiro-conjugation in the sense that we use, but as a transition state property, has also been proposed for so-called coarctate reactions (e.g. 4)11 for which a 4n s-electron Möbius-like transition state requires the central atom to be tetrahedral (and to be square planar for a 4n+2 process). We have recently suggested12 that this concept of coarctate aromaticity could be extended to minima in the potential energy surface, where the coarctate atom would a four-coordinate bromine at the intermediate for Br+ transfer reactions of alkenes. Combining the concepts ilustrated by 1-4 led us to speculate about the generality of a novel class of aromatic molecule formed by conjugated rings sharing a mutual atom rather than a mutual bond, and whether any aromaticity manifested might be a local phenomenon associated just with each ring, or an intrinsic property of the combined coarctate system.11 Here we report model calculations investigating these propositions.
Computational Procedure: Molecular structures were optimised using Gaussian9813 at the closed shell B3LYP/6-31G(d) level. Calculations of NICS values14 were conducted at either the ring centroid only for rings with C2 distortions, or at the ring centroid and 1Å above the ring centroid (NICS(0) and NICS(1) respectively) for the essentially planar rings. Coordinates are available via the supplemental data at http://www.rsc.org/...
Results and discussion:
The systems 5-10 were designed to explore both formal
4n pelectron occupancy in the ring A
component of the spirocyclic system (systems 5,
6, 9, 10) and 4n+2 occupancy (systems
7, 8). Likewise ring B can have either 4n+2
(5-8) or 4n (9,10) p
occupancy as either a phosphacarbocycle (6, 8,
10) or a cyclophosphazene (5, 7 or
9) ring. Of these models, systems 5 and 7
have close analogy to known molecules8. Thus
analogues of 5 differing only in dibenzo substitution of
ring A and in tris rather than mono substitution at each
phosphorus are known, and likewise for 7. Ab
initio B3LYP/6-31G(d) energies and geometries are listed in
the Table.§
To avoid concealing the electronic conformational preference of the ring, we calculated the sterically unhindered parent ring systems in most cases. The ring substituents R=H and also R=F were studied, the latter having being shown to enhance Möbius aromatic character in particular.4-6 With 5 and 6, X=O, only a C2 symmetric conformation could be located but for R=F,X=NF, two conformations could be located, that with C2 symmetry being of lower energy than for Cs symmetry. This result contrasts with the conformational surface found6 for the related heteropine ring systems 11, for which Cs symmetric conformations are invariably lower in energy than the C2 Möbius forms. This reinforces the interpretation that the C2 symmetry arises from Möbius electronic contributions rather than originating entirely from steric factors.
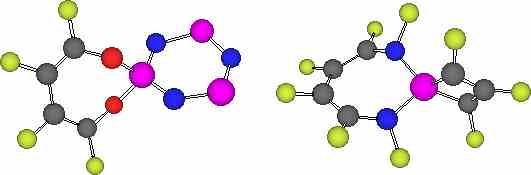
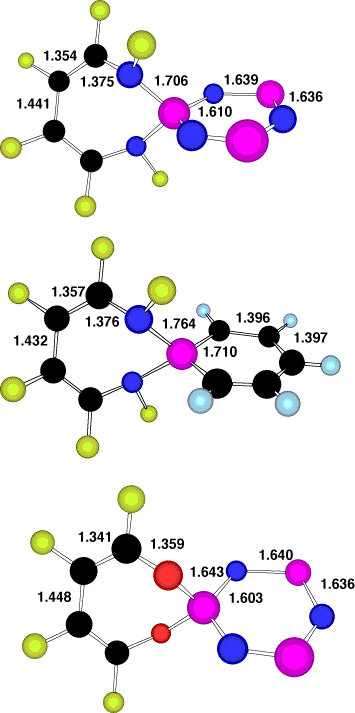
The NICS values for 5 and 6 show essentially non-aromatic characteristics for ring A with R=H, X=NH,O true also for the dibenzo system substituted analogue of 5, X=O, R=benzo. We have noted previously4 that this ring system can exhibit a continuum between 8-p planar anti-aromaticity and 8-p non-planar Möbius aromaticity. The latter is enhanced by fluorine substituents, and accordingly with 6, X=NF, R=F, ring A now exhibits an aromatic negative NICS(0) value (compared to benzene for which NICS(0) ~-10 ppm). Where ring B is a phosphabenzene, it too shows modest aromatic character, as reported previously by Schleyer and co-workers1. The cyclophosphazene system however appears only very mildly aromatic (Table) using the NICS(1) criterion (and slightly anti-aromatic using the NICS(0) probe), a property not previously reported, and rather surprising given the essential planarity of this ring and the equivalence of all the ring bonds (Figure 1). This may in part be related to the ability of the three nitrogen ring atoms in this ring to bear negative charge, favouring contributions such the dipolar resonance form shown for 5. Computed bond lengths support this interpretation, the X-P values for 5 being 1.706/1.643Å (X=NF/O) compared with those in 6 of 1.764/1.698Å (X=NF/O) (Figure 1).
Finally in this category of ring, we note that the (experimentally unknown) ring system 6, X=S, R=F shows both rings to be significantly aromatic and the aromaticity is even more prominent with the arsenic analogue15 of 6, R=F, X=O.
Systems 7 and 8 are the formal 4n+2 homologues of ring A. With X=O, ring A is essentially planar with only a small distorsion to Cs symmetry, in agreement with observed structures16. NICS values show it to be aromatic. As before, the cyclophosphazene ring B is non aromatic, whereas the phosphabenzene ring B is clearly aromatic. With X=NF, pyramidalisation of the nitrogen occurs in either cis or trans fashion. In each case, the cis form was of lower energy, and showed Cs symmetry, whereas the trans form had no symmetry, possibly due to a Jahn-Teller like distorsion of the anti-aromatic Möbius form of a 4n+2 ring.
The pair of systems 9 and 10 reveal a Möbius conformation and NICS(0) values corresponding to aromaticity for ring A, and in the case of 6, R=F, X=O, NF, also a C2 Möbius non-planar aromatisation for the phosphacyclobutadiene ring B, especially when it too is fluorinated (R'=F). As before, the O-P length for 9 (1.638Å) is shorter than that for 10, R'=H (1.661Å) or 10, R'=F (1.640Å), indicating contributions from ionic forms.
Comparing the pair 5/9 , X=O, R=F reveals the NICS(0) value for ring A to vary little (-4.5 to -5.0 ppm) whilst the essentially planar ring B changes from a formally 4n+2 p aromatic to a 4n p planar anti-aromatic system, reflected in a change of the NICS(1) value from -2.3 to 0.2 ppm. The NICS(0) value for ring B, which at the ring centroid also includes more paratropic contributions from the s framework13 reveals a small opposing trend. Similar trends occur for 5/9, X=NF, R=F. The pair 6/10, X=O,R=F shows similar constancy for ring A (-5.9 to -4.6) whilst ring B varies from -7.9 to -17.2 ppm (changing from a formally 4n+2 p aromatic to a 4n p Möbius aromatic). An alternative way in which to count the p electrons is as a single ring system linked via a coarctate phosphorus atom involving some degree of d-p orbital participation, analogous to the transition state coarctic model proposed by Herges and coworkers.10 For this model, 5/6 are 14 (4n+2) electron systems whereas 9/10 are 12 (4n) p electron systems. The NICS values imply that the aromaticity is in fact localised to each ring, and does not follow coractate rules, with the obvious caveat that measuring NICS values at individual ring centroids may not be an appropriate measure of overall coarctate aromaticity.
ConclusionsWe conclude that spiro-conjugated rings can exhibit both conventional Huckel p aromaticity and for systems such as 5 of which there are known analogues, more unusual Möbius contributions to the aromaticity in one ring. With phosphorus as the coarctate atom, each ring appears to sustain aromaticity associated with its own 4n or 4n+2 p-electron count. Spiro-aromatic systems where the aromaticity of each ring is more strongly coupled via the spiro-atom remain an intriguing possibility, as do systems where the spiro atom could reveal square planar rather than tetrahedral geometry.
§Coordinates for both computed and experimental geometries are available via the ESI at http://www.rsc.org/...
| Table 1. B3LYP/6-31G(d) Calculated energies (Hartree) and NICSa Values (ppm) for 5-10 | |||||
|---|---|---|---|---|---|
| Substituents | Energy | NICSa | Substituents | Energy | NICSa |
| 5, X=O, R=H | -1493.59245 | 1.0 (-2.2)/-0.4 | 5, X=NH, R=H | -1453.84743 |
1.9 (-1.8)/-0.3 |
| 5, X=O, R=benzo | -1800.91060 | 1.5 (-1.7)/1.2 | |||
| 5, X=O, R=F | -1890.49767b | 0.9 (-2.3)/-5.0 | 5, X=NF, R=F | -2049.05166 (2.8)c |
1.5 (-2.0)/-9.6 |
| 6, X=O, R=F | -1237.02783b | -7.9 (-7.7)/-5.9 | 6, X=NF, R=F | -1395.58808 (7.0)c |
-7.7(-7.6)/-11.6 |
| 6, X=S, R=F | -1882.94836 | -5.3 (-5.6)/-9.5 | |||
| 6, X=O, R=F, Arsenicd | -3129.40995 | -7.6 (-8.0)/-7.8 | 6, X=NF, R=F Arsenic | -3287.97290 | -7.3 (-7.8)/-12.2 |
| 7, X=O, R=F | -1614.62798 |
0.3 (-2.8)/-7.8 | 7, X=NF, R=F |
-1773.19255 (5.2)e |
2.4/-9.2 |
| 8, X=O, R=F | -961.16074 |
-8.8 (-8.6)/-7.1 | 8, X=NF, R=F |
-1119.72821 (4.0)e |
-7.7 (-7.5)/-9.2 |
| 9, X=O, R=F | -1494.32476 |
-0.9 (0.2)/-4.5 | 9, X=NF, R=F |
-1652.88226 |
0.4 (0.6)/-8.6 |
| 10, X=O, R=F, R'=H | -1159.51846 |
2.6/-4.8 |
10, X=NF, R=F, R'=H |
-1318.07670 |
5.2/-8.3 |
| 10, X=O, R=F, R'=F | -1457.18499 | -17.2/-4.6 | 10, X=NF, R=F, R'=F |
1615.74390 (1.3)f |
-16.2/-8.3 |
aNICS(0) value at Ring B centroid (NICS(1) value 1Å above ring B centroid)/NICS(0) value at ring A centroid. bGeometry optimisation converges to C2 symmetric conformation. cRelative energy (kcal/mol) of Cs symmetric conformation. dAs replacing P. eRelative energy (kcal/mol) of anti-conformation, with no symmetry. fRelative energy (kcal/mol) of chiral diastereoisomer.