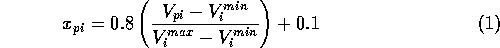Stuart
M. Green
Stuart
M. Green
 and Henry S. Rzepa
and Henry S. Rzepa
Howard B. Broughton, Stuart
M. Green and Henry S. Rzepa
Neuroscience Research Centre,
Merck Sharp and Dohme, Terlings Park, Harlow, Essex CM20 2QR
Department of Chemistry, Imperial College,
London SW7 2AY
To whom all correspondence should be
addressed. Current address: Center for Molecular Design, Campus Box
1099, Washington University, St. Louis, MO 63130-4899. Email:
stuart@wucmd.wustl.edu
 . Using MEPs determined with a variety of semiempirical
and ab initio methods, the predictive power of the trained
network was found to be sensitive to the quality of the basis set. The
network was also trained to predict the proton affinity (PA) and p, both individually and combined, for a
series of pyrazole MNDO MEPs.
. Using MEPs determined with a variety of semiempirical
and ab initio methods, the predictive power of the trained
network was found to be sensitive to the quality of the basis set. The
network was also trained to predict the proton affinity (PA) and p, both individually and combined, for a
series of pyrazole MNDO MEPs.
Neural networks are now finding regular application to a variety of chemical problems: spectroscopy[2][1], prediction of protein structure[6][5][4][3] and water binding sites[7], synthetic analysis[10][9][8], solvation studies[11]. This rapidly growing field has already spawned two reviews, by Burns and Whitesides[12], and Gasteiger and Zupan[13]. The realm of quantitative structure-activity relationships (QSAR) has seen a great deal application of such neural paradigms[18][17][16][15][14] as they make an obvious alternative to the multivariate statistics traditionally used in Hansch analysis.
In preliminary studies we successfully trained a neural network with a
series of imidazole MEPs (AM1 derived) in attempt to predict histidine
p perturbation in triosephosphate isomerase[19]. Such a
regime uses the neural network to derive a three-dimensional
quantitative structure-activity relationship (3D-QSAR) - inputs to
the network are sourced directly from grid points of the MEP (freeing
us from the restrictions imposed by substituent parameters). The
network is then trained to map these points to the molecular property
(p). Statistical techiniques such as comparative molecular field
analysis (CoMFA[20]) have also been developed to perform 3D-QSAR,
but these methods presuppose some form of non-parametric[21] or
more restrictive parametric[20] relationship between individual
field points and the observable quantity. Neural networks offer an
alternative where no assumption is made about the relationship between
property and field; instead this is derived during the process of
training the network.
To test the 3D-QSAR abilities of the neural network further, this paper
details the results of using different basis sets or Hamiltonians for
deriving the MEP in the imidazole p case. The results of training
pyrazole p and/or proton affinity (PA) against MNDO MEPs are also
discussed.
All structures were optimised using either MOPAC[22] or Gaussian[23] for the respective semiempirical
method (AM1, PM3 and MNDO, using EF and PRECISE) or
basis set (STO-3G, 3-21G and 6-31G*). Each optimised molecule was then
aligned such that atom 1 (Figure 1 and
Figure 3) was placed at the origin,
with atom 2 extending out along the positive X-axis and atom-3 in the
XY-plane. Where atoms 1, 2 and 3 are consistently defined for all. The
MEP was then evaluated, with each respective method, on a 1.5 Å
grid excluding points that were within the van der Waals radii and
those which were more than 6 Å from the closest atom .
.
For suitable input to the neural network only grid points common to all molecules in either imidazole or pyrazole set were used. Each MEP point was then scaled such that the value presented to the network lay in the range 0.1 to 0.9 (in order to prevent network saturation).
Where 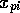 is the scaled input for
molecule
is the scaled input for
molecule 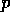 at grid location
at grid location 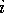 ,
, 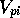 is
the MEP value,
is
the MEP value, 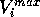 and
and 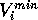 are the respective maxima and minima for
all molecules at the MEP point . The p values to
which the inputs would be trained against were also similarly
normalised.
are the respective maxima and minima for
all molecules at the MEP point . The p values to
which the inputs would be trained against were also similarly
normalised.
The neural network used in this study was of the backpropagation
type[26][25]
with one hidden layer of 10 neurons and a final layer with a one
neuron, for single property prediction, or two, when combined
properties are used. All neurons had a sigmoidal activation function
and a single bias term (fixed input of 1.0). Initially, all neuronal
weights were randomly set (uniform distribution) to values between
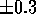 . The scaled MEPs
were trained to map to their respective properties in `batch mode',
ie. all patterns were presented and the network updated with
the sum of errors. Network updates were performed using a fast
adaptive learning algorithm (superSAB[27]). This method was found to be much
stabler and require far less iterations to achieve the required degree
of training than the traditional generalised delta rule[25].
. The scaled MEPs
were trained to map to their respective properties in `batch mode',
ie. all patterns were presented and the network updated with
the sum of errors. Network updates were performed using a fast
adaptive learning algorithm (superSAB[27]). This method was found to be much
stabler and require far less iterations to achieve the required degree
of training than the traditional generalised delta rule[25].
Due to the initial random weights the network cannot be guaranteed to produce the same results for every random seed, hence many differently seeded trainings were used for each crossvalidation step with all predictions included in the subsequent analysis.
QSAR with
MEPs. A collection of small, conformationally invariant imidazoles of
varied substitution were selected (Table 1), these covered a range of 10 p units. The geometry of each of these 26
imidazoles was optimised and MEPs calculated for all methods
described. A leave-one-out crossvalidation analysis was then
performed for all 26 imidazoles training five differently seeded
networks to map MEP to p for 25 of the
imidazoles, then predicting the p of
the excluded imidazole (Figure 2).
Two molecules (5NO -1Me and
5F-1Me) are expressed as outliers independent of the MEP derivation
methods, this is due to the lack of 5-substituted moieties and the
network is relatively uneducated about these types of species. With
the semiempirical methods and STO-3G the common outliers are all
molecules containing chlorine suggesting a poor electrostatic
description of chlorine by these methods due to an incorrect
definition of the atomic core[28].
-1Me and
5F-1Me) are expressed as outliers independent of the MEP derivation
methods, this is due to the lack of 5-substituted moieties and the
network is relatively uneducated about these types of species. With
the semiempirical methods and STO-3G the common outliers are all
molecules containing chlorine suggesting a poor electrostatic
description of chlorine by these methods due to an incorrect
definition of the atomic core[28].
Overall,
![]()
the best performing networks were those trained with the high level ab initio derived MEPs, although there is little improvement (but considerable effort) in going form 3-21G to 6-31G*. The poor performance of PM3 is to be expected due to the poor parameterisation of nitrogen[29].
In previous comparisons of MEP derivation methods[31][30], workers have assumed those determined for the highest feasible level of theory were the most `realistic' and used this level as a standard. From this work with imidazoles one can suggest an order of how realistic the techniques are in producing MEPs by correlating them with real experimental data, rather than via comparison with a presumed standard.
and PA QSAR with
MEPs.Neural networks were also trained to map the MEP to the p and PA of a series of pyrazoles. Recent
experimental (fourier transform ion cyclotron resonance spectroscopy)
and theoretical (6-31G geometries and energies) work has demonstrated
that for the tautomerism of NH pyrazoles (Figure 3) the equilibrium constant 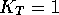 [32] ie. 3- and 5-substituted
moieties are inseparable, hence their p's and PAs are identical and both tautomers should be
considered equally.
[32] ie. 3- and 5-substituted
moieties are inseparable, hence their p's and PAs are identical and both tautomers should be
considered equally.
Proton affinity data[32] for 39
pyrazoles was used, 10 of these had equivalent tautomers, so a total
of 49 pyrazoles were considered. The geometries of these were
optimised and the MEP evaluated as before for the imidazoles but only
at the MNDO level. A complete leave-one-out crossvalidation study for
each pyrazole was carried out for five differently seeded
networks. The results of this study are shown in Figure 4(a). A much larger dataset of 58 pyrazole
p's was available[33], of these 33 had equivalent tautomers
making an overall set of 91 pyrazoles. Exactly the same approach was
used as in the proton affinity study, results in Figure 4(b).
Training is not restricted to the mapping of only one property at
a time, any number of outputs can be included and the network trained
to predict many things at once. Of the pyrazoles considered 21 are
coincident between the p and proton
affinity sets. Thus, these were used in a crossvalidation study but
with training based on two outputs. For comparison purposes PA and
p were also trained independently for
the 21 pyrazoles. The results of this combined training (Table 5) are rather poor, the crossvalidations
are nowhere near as good for those observed previously. This is not a
failure of the network to cope with two outputs but just due to a lack
of training data. Independent trainings on just the 21 pyrazoles
against proton affinity or p yield
almost identical results, hence the training of multiple properties is
not detrimental to predictive performance.
We have demonstrated the utility of neural networks in the correlation of complex quantum mechanically defined features (MEPs) with experimentally determined parameter(s) for a given molecular series. The neural network, far from being the blind pattern classification method, shows the ability to generalise as it remains sensitive to the relative level of theory. Further studies based on trainings using alternative grid-based molecular descriptors and other properties (eg. biological activities) will hopefully expand this technique fully within the realm of 3D-QSAR.
We thank Merck Sharp and Dohme Research Laboratories and the SERC for financial support (to S.M.G.).
Alternative grid
definitions and numbers of neurons in hidden layer have been tested
but this prescription was found to be best in a trade-off between
performance and computation time[24]
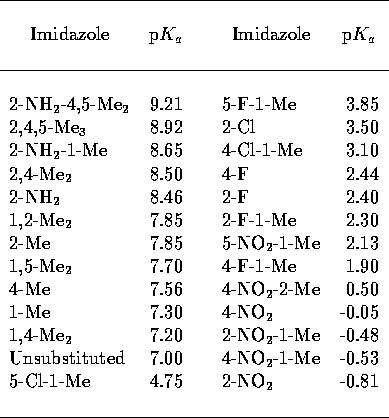
Table 1: Substituted imidazole p[34]

Table 2: Crossvalidation statistics for mapping of MEP to p for 26
imidazoles. Each analysis based on all five different trainings.
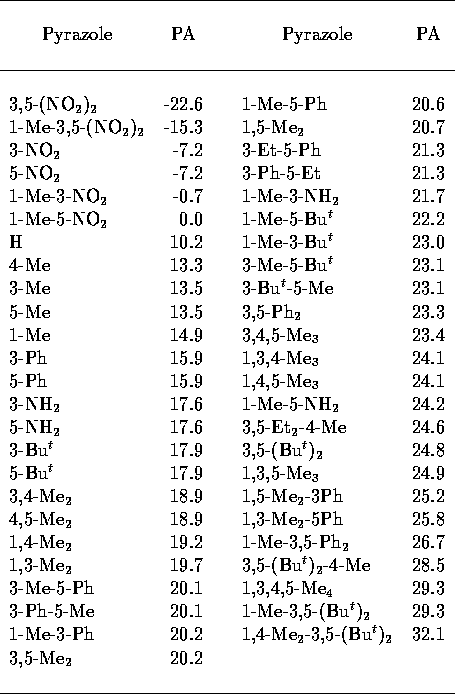
Table 3: Experimental pyrazole
PA[32].
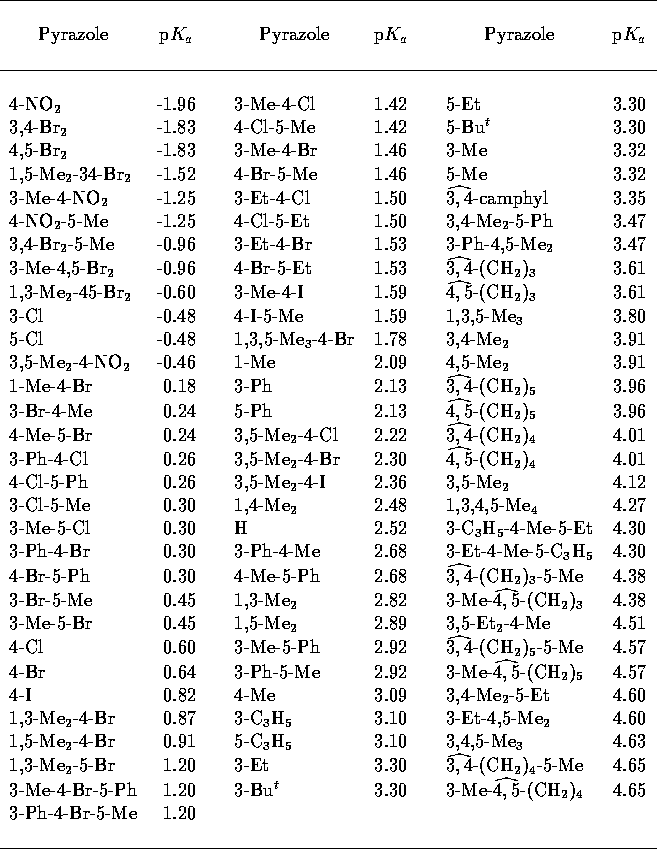
Table 4: Experimental pyrazole
p[33].
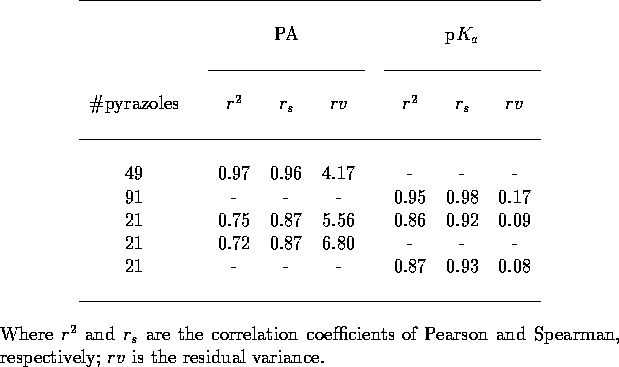
Table 5: Crossvalidation
statistics for pyrazole MEP training. All five different leave-one-out
crossvalidations included in each analysis.
Figure 1: Imidazole atom numbering.
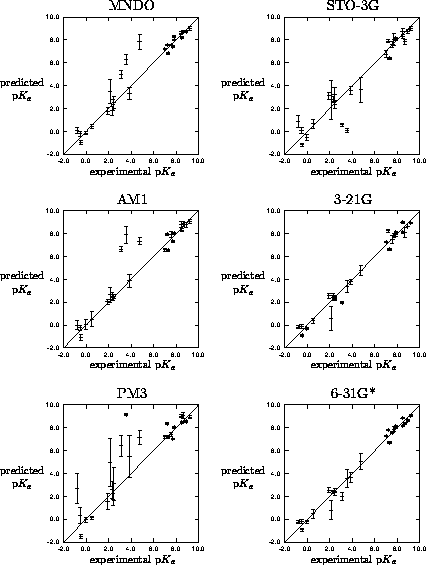
Figure 2: Leave-one-out crossvalidation results for
mapping of MEP to p for 26 imidazoles. Predicted p is mean of
five independent trainings with error bar as the standard deviation.
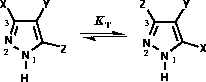
Figure 3: Pyrazole tautomerism and atom numbering.
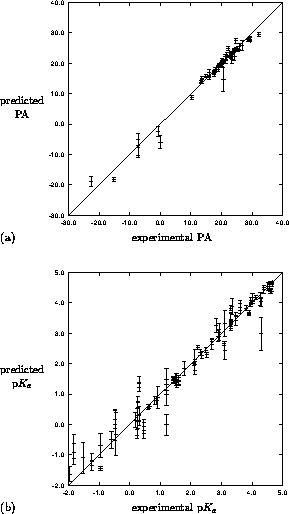
Figure 4: Leave-one-out crossvalidation results for
mapping of MEP to (a) PA for 49 pyrazoles and (b) p for 91
pyrazoles. Predicted property is mean of five independent trainings
with error bar as the standard deviation.