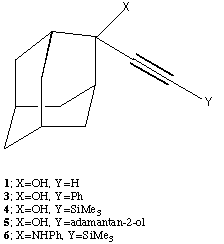
Department of Chemistry, Imperial College, London, SW7 2AY.
Summary. Four analogues of 2-ethynyl-adamantan-2-ol (1) have been synthesised and their crystal structure investigated. All four exhibit distinctive intermolecular hydrogn bonding patterns, but only the 2-phenylamino system (6) revealed the presence of π-facial hydrogen interactions similar to those found in 1.
There is much current interest in probing the diversity and limits of hydrogen bonding interactions to π systems such as phenyl rings and double and triple bonds. We have previously identified[1] a small number of molecules for which there is crystallographic evidence for relatively strong OH...π interactions to double or triple bonds. Of these, the most intriguing is the structure of 2-ethynyl adamantan-2-ol 1, which independently has correctly been identified[2] as containing both intermolecular OH...π and C-H...O interactions.
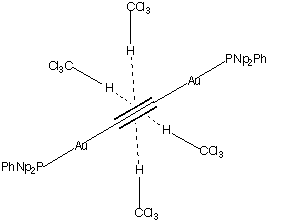
Mingos, Muller and Williams[3] have recently reported a remarkable structure for the Gold (I) ethynyl complex 2, which shows four significant orthogonally oriented π...Η-CCl3 interactions to each central triple bond.
As part of our program[4] of studying π-facial interactions, we wished to establish the structural tolerance of such interactions, and in particular whether simple analogues of 1 might also demonstrate this effect. We report here the syntheses of four related ethynyl adamantane derivatives 3-6, the cyanohydrin derivative 7 of adamantanone and the crystal structures of 3, 4, 6 and 7.
Experimental Details.
2-Phenylethynyladamantan-2-ol: (3): Phenyl acetylene (0.43ml, 0.4g, 3.9 mMol) was dissolved in tetrahydrofuran (2 ml) and a solution of butyl lithium in hexanes (2.45 ml of a 1.6M solution, 3.9 mMol) was slowly added. Then a solution of adamantanone (0.52g, 3.46 mMol) in THF (2ml) was added slowly. The reaction mixture was stirred at room temperature for one hour before the addition of water (1ml, 55mMol). The reaction mixture was added to diethyl ether (100 ml), and this was then dried with magnesium sulphate. Removal of the solvents gave a yellow oil which was mixed with n-Hexane and was recrystallized to give a white solid (33 mg, 3.3%). Removal of the hexane from the mother liquor gave a yellow solid which was recrystallized from hexane to give a white crystalline solid ( 0.247g, 0.979 mMol, 28%) found C 85.48%, H 8.60% (calculated 85.7%, H7.9%). m.p. 96-98oC. Crystals were grown by the slow evaporation of solvent from a dilute hexane solution.
2-Trimethylsilylethynyl adamantan-2-ol (4): Trimethylsilylacetylene (0.58ml, 0.4g, 4.07 mMol) was mixed with tetrahydrofuran (10 ml), and the solution cooled to -78oC. Butyl lithium in hexanes (3 ml. 3.5 mMol) was added slowly with stirring. The mixture was stirred for one hour at -78oC before the addition of adamantanone (0.5g. 3.33mMol) dissolved in THF (15 ml). This was stirred for ten minutes at -78oC before being allowed to warm to room temperature, stirred for two hours and excess ammonium chloride added as a quench. The ammonium chloride the solution was filtered, and the solvent removed. Extraction with hot pentane followed by filtration and evaporation gave a heavy oil which slowly crystallized to give a waxy solid ( 0.508 g. 2.05 mMol. 61%). Crystals suitable for X-ray analysis were formed by the slow evaporation of solvent from a solution in hexane.
Bis(adamantan-2-ol) ethyne (5): Purified acetylene gas was bubbled rapidly through a well stirred solution of butyl lithium (7.7 ml of a 1.17 M solution in hexane, 9.01 mMol) mixed with diethyl ether (10 ml, sodium dried) for two hours at room temperature. The suspension was stirred under nitrogen gas for two hours to allow the disproportionation reaction to occur, before the addition of adamantanone (1.3 g, 8.7 mMol) dissolved in THF (24 ml). The mixture was stirred for one hour before being heated under reflux for 16 hours. After cooling, the now clear orange liquid was treated with water (0.2 ml 11 mMol), the ether layer dried with anhydrous sodium sulphate and filtered. The solvents were removed to give an orange solid (2 g), and recrystallized from a mixture of dichloromethane (50 ml) and hexane which was cooled to -78oC to precipitate out a white microcrystalline solid (0.727g, 2.23 mMol, 52 %). A small sample was purified by vacuum sublimation (0.6 Torr. 180oC), m.p. 304-306. Found C 80.91%, H 9.17% (calculated C 81%, H 9.2 %). 1H: δ1.95, 13C: 89.3, 72.4, 39.0, 37.7, 35.6, 31.6, 26.9, 26.8.
2-Trimethylsilylethynyl 2-phenylamino-adamantane (6): A solution of trimethylsilylacetylene (0.65g, 6.7mMol) in dry THF (90ml) was cooled to -78[[ring]]C before the addition of 5.6ml of 1.6M BuLi (8.96mMol, 1.3eq, in hexane), followed by stirring at room temperature for a further two hours under nitrogen. Separately, a 250 mL three-necked round-bottom flask fitted with a soxhlet was purged with nitrogen and charged with 150ml dry toluene, 1g adamantanone (6.65mMol), 0.65ml aniline (7.2mMol) and a trace of tosic acid (8mg) before heating under reflux until no hydrogen development could be observed in the thimble of the soxhlet. This second solution was added dropwise to the lithium acetylide solution producing a Bordeaux-red product, which was allowed to boil under reflux for 67h to give a dark brown solution, passed through a Silica column (Petrol/Ethyl acetate=2.5:1) and the collected fractions dried over K2CO3. Evaporation of the solvent afforded 2g of a brown oil from which orange crystals could be collected, mp 59-65[[ring]]C. Recrystallisation in an aqueous methanol solution gave white crystals, m.p. 73[[ring]]C. 1H NMR δ 0.7-2.25 (Adamantyl-protons); 1.59 (s, 9H): TMS, 3.6 (s, 1H):NH; 7.00 (t, 1H), 7.02 (d, 2H), 7.03 (t, 2H): aromatic set.13C NMR δ 145.4, 128.5, 118.0, 116.5, 109.7, 90.1, 77.2, 77.0, 76.74, 57.5, 37.9, 35.6, 34.5, 31.5, 29.7, 27.04, 26.6. MS m/z 323, 250, 231, 97, 73. IR (Nujol) 3364.5 (N-H), 2157.1 (-C[[equivalence]]C-), 841.1 (-Si(CH3)3) cm-1.
2-Cyanoadamantan-2-ol (7): Adamantanone (0.45 g. 3.0 mMol) in methanol (2 ml) was treated with concentrated sulphuric acid (0.5 ml) and the solution was allowed to cool to room temperature before the addition of sodium cyanide (1.0 g, 20 mMol) dissolved in water (2.2ml). The reaction mixture was heated under reflux for one hour before being stirred at room temperature for 90 minutes. Diethyl ether was added to the reaction mixture and this was then stirred before being filtered. The solid salts in the reaction flask were washed repeatedly with diethyl ether. After filtration these washings were combined with the filtrate.
The combined extracts were washed with a saturated sodium chloride solution, and then with water before being dried with magnesium sulphate. After filtration the ether extract had its solvents removed in vacuo to give a pale brown solid product. The crystals from the diffusion of hexane vapour into a solution in ethanol were suitable for X-ray crystallography. (0.337 g, 1.9mMol, 63%). IR 3401 sharp, 2909, 2859, 2240, 1574, 1453, 1354, 1118, 1102, 1067, 1029, 995, 922 and 737.
Crystallographic Details: A summary of the crystal data, data collection and refinement parameters for each compound is given in Table 1. Crystals of the symmetric bis-ethynyl-adamantol system 5 were found to have large unit cell parameters indicating the presence of at least 16 crystallographically independent molecules in the asymmetric unit. Further structural studies of 5 were thus considered to be impracticable. However, such a large number of independent units is highly unusual and indeed noteworthy. Attempts to produce isomorphs by growing crystals from other solvents all gave this same result. Data for all other compounds were measured using graphite monochromated Cu-Kα radiation using ω-scans. Data for 3 were collected on a Siemens P4/PC diffractometer whilst those for the remaining compounds were measured on a Siemens P4 rotating anode instrument. Data were corrected for Lorentz and polarisation factors, but not for absorption. The structures were solved by direct methods and the major occupancy non-hydrogen atoms were refined anisotropically. In 6 the phenyl ring in each of the partial occupancy molecules was refined as an idealised rigid body. The positions of all the major occupancy O-H and N-H hydrogen atoms were located from DF maps. The hydroxyl hydrogen atoms were refined isotropically, U(H) = 1.2Ueq(O), subject to O-H distance constraints. The positions of the remaining hydrogen atoms were idealised, assigned isotropic thermal parameters, U(H) = 1.2Ueq(C/N), [U(H) = 1.5Ueq(C) for CH3 groups] and allowed to ride on their parent atoms. In structure 6, the relative occupancies of the two molecular orientations were determined by refinement of a free variable tied to the occupancy parameters. Refinements were by full matrix least squares and based on F for 3, 4 and 7, and on F2 for 6. Computations were carried out using (for 3, 4 and 7) the SHELXTL 4.2 program system and (for 6) the SHELXTL 5.03 program system, on a combination of 486 PCs and a Silicon Graphics Iris/Indigo workstation.
Fractional atomic coordinates, thermal parameters and bond lengths and angles for all the structures have been deposited with the Cambridge Crystallographic Data Centre. The crystal structures of 3, 4, 6 and 7 are available on the World-Wide-Web system via the electronic version of this paper in the form of "hyperactive" molecules[5] and available via the URL http://www.ch.ic.ac.uk/rzepa/RSC/P2/5_XXXXX.html The searches of the Cambridge structural database were undertaken using Version 5.6 of the Quest program system and database,[6] containing 120123 entries.
Results and Discussion: Compound 3
crystallises with four crystallographically independent molecules in the
asymmetric unit, each adopting an essentially identical conformation. The bond
lengths within each molecule are normal with the acetylenic bond lengths in the
range 1.183(5) to 1.197(5) Å. The molecules pack to form hydrogen
bonded cyclic tetramers which have approximate, non-crystallographic, C4
symmetry (Figure 2). Each O-H group acts both as a donor and as an
acceptor of a hydrogen bond with O...O, H...O and O-H...O distances and angles
in the ranges 2.79 to 3.07, 1.96 to 2.28 Å and 155 to 165deg.
respectively.+ Supplementing these hydrogen bonds are intermolecular
T-type aromatic-aromatic edge-to-face interactions involving all four phenyl
rings within each tetramer. The aromatic-aromatic centroid-centroid distances
are in the range 4.90 to 5.02 Å (values typical for this type of
interaction). The tetramers in turn are packed such that one of the methylene
C-H protons in each of the four independent molecules [attached to C(10)] is
directed towards the centroid of an aromatic ring, approximately diametrically
opposite to the edge-to-face interaction present within the tetramer (vide
supra, Figure 3). The H...centroid distances are in the range 2.70 to
2.84 Å and the H...centroid...centroid angles are in the range 162
to 171deg.. These intermolecular stabilising interactions are cumulative and
extend throughout the crystal with each individual molecule being involved in
six intermolecular interactions.
Concluding that the phenyl substituent in 3 is likely to conjugate with
the alkynyl group and hence decrease the available electron density for
π-facial interactions, we turned to 4, where the electropositive
nature of the silicon was expected to enhance the basicity of the triple bond.
The geometry
is unexceptional with an acetylenic bond length of
1.201(8) Å [C(11)-C(12)]. The principal point of interest in this
compound is in the packing of the molecules in the crystal. In contrast to the
tetrameric motif seen for 3, molecules of 4 are arranged to form
hydrogen bonded cyclic hexamers with crystallographic S6 symmetry, as
shown in Figure 5. The six oxygen atoms of the hydrogen bonding array are
arranged in a cyclohexane-like chair conformation (Figure 6). The O...O
and H...O distances, and the O-H...O angles are 2.77 and 1.90 Å, and
159deg. respectively.[[daggerdbl]] Within each cyclohexane-like
array of oxygen atoms, the O...O...O angles are 105deg.. There are no
intermolecular approaches of less than 3 Å of any C-H protons to the
acetylenic triple bond.
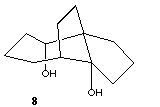
Searches of the Cambridge structural database revealed that both cyclic tetrameric and hexameric arrangements of alcohols are not uncommon,[7] the latter exhibiting a distinct preference for the chair conformation. We note in passing one unusual diol 8,[8] which forms a cyclic hexamer comprising six intra and six intermolecular hydrogen bonds in which 24 atoms are involved in the ring.
We also found that cyclic trimers are much rarer. 1-Methylindole-5,6-diol 9 is a typical example of this cyclic form, showing a triangular arrangement of oxygen atoms with the three hydrogens not deviating far from the axis of the O...O atoms, combined with bifurcation at the three hydrogens. In the original report of both 9 and other systems,[9] the existence of intermolecular and intramolecular hydrogen bonds was noted but no comment was made on the unique trimeric features of these systems.
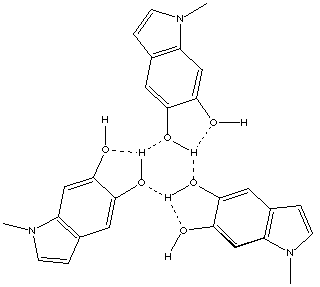
9
Compound 6 crystallises with a geometry corresponding to that
illustrated in Figure 7.
An initial data set for a crystal cut from an
aggregate formed from the melt produced a well defined molecular unit, but
would only refine to a residual R = 0.076. A difference
electron density map, however, still contained peaks of up to
0.7 eÅ-3 which appeared to suggest an alternative
overlapping molecular orientation. These peaks could not be resolved to give a
discernible complete molecule. A second data set collected for a crystal of
well defined morphology obtained by crystallisation from aqueous methanol still
produced a structure solution with unresolvable residual electron density
`ghosting'. A third data set collected at 123 K still contained ghosting.
In this instance, however, it was possible for it to be resolved to provide a
complete partially overlapping alternative orientation of the whole molecule,
though with some atom sites almost coincident (Figure 8). This arrangement
corresponds to the partial superimposition of the molecule upon its enantiomer.
The only conformational feature of note is a near coplanarity (7deg. torsional
rotation about N(1)-C(26)) of the quaternary adamantyl carbon atom C(1), the
amino nitrogen atom N(1) and the aromatic ring plane. Accompanying this
geometry is a directing of the ortho aromatic proton attached to C(21)
towards the acetylenic triple bond [C(11)-C(12) 1.209(3) Å]
such that the hydrogen atom lies 2.54 Å from C(11) and
2.69 Å from the centre of the triple bond (see Figure 7). The
values are similar to the distances found for the analogous interactions
observed in compound 2. The vector linking H(21A) to the centre of the
acetylenic bond is inclined by 70deg. to the triple bond axis.
Centrosymmetrically related pairs of molecules are oriented such that the amino
proton in one molecule is positioned over the acetylenic bond in the other
and vice versa (see Figure 9). The H...triple bond centroid
distance is 3.0 Å.[[section]] The associated H...C(11)
and H...C(12) distances are 3.19 and 2.93 Å respectively. The
H...triple bond centroid vector is inclined by 77deg. to the triple bond axis.
The angle subtended by the two H...centroid vectors is 119deg. (q.v.
90deg. for the corresponding angle in 2).
The NH...triple bond interaction exhibited weakly in 6 is nevertheless rare. A search of the Cambridge database revealed only two other compounds, the neutral 5-(2-propynyloxy)-2'-deoxyuridine[10] and the ionic hydrazinium tetraethynylborate hydrazine[11] which show similar intermolecular NH...π triple bond interactions. No NH-π alkyne interaction has yet been identified with an interaction as strong as those found in the hydroxy system 1.
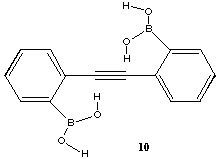
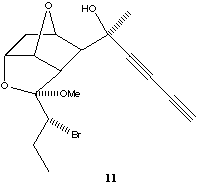
Compound 1 itself is not as unique as we first thought. For example, Wallis[12 has shown 10 to have a OH...π interaction. Calculations[13] on this system revealed the relatively long OH...C distance of 2.52Å to the nearest carbon to be due to the participation of an additional bifurcated OH...O intermolecular hydrogen bond, subsequently verified in the crystal structure. We have shown that the synthetic intermediate 11[14] also contains an example of an intramolecular OH...π interaction with an intermolecular bifurcation to an ether oxygen (rOH...C 2.18Å, ROH...O 2.05Å).
To complete this study, we found that the crystal structure of the cyanohydrin
of adamantanone (7) had not hitherto been determined.
It crystallises
with two crystallographically independent molecules in the asymmetric unit in a
non-centrosymmetrical polar space group (Figure 10). The two independent
molecules are linked via an O-H...N hydrogen bond [O...N 2.89,
H...N 2.00 Å, O-H...N angle 168deg.].[[daggerdbl]]
Pairs of molecules are in turn linked by a further O-H...N hydrogen bond
[O...N 2.89, H...N 2.00 Å, O-H...N angle 170deg.] forming
sinuous chains that extend in the crystallographic c direction
(Figure 11). Adjacent chains in the b direction are cross-linked
via weak C-H...O interactions between one of the methylene protons
[attached to C(10)] in one chain and an oxygen atom O(1) in the next
[C...O 3.49 Å]. Although this interaction is at the margin of
significance [H...O 2.57 Å, C-H...O angle 159deg.] the geometry
of approach to the oxygen centre is intermediate between tetrahedral and
trigonal--the H...O-H and H...O-C angles are 120 and 117deg..
Conclusions. Our results show that heteropolar interactions between OH or NH groups and triple bonds form relatively rarely, and that the more conventional O-H...O interactions will dominate whenever possible. Surprisingly, C-H...C[[equivalence]]C interactions were also detected in the systems studied, and overall the diversity of the intermolecular interactions is far more subtle than normally presumed.
We thank the Wolfson Foundation for equipment and computing grants.
Footnotes.
+ The O-H distances were constrained to 0.85 Å.
[[daggerdbl]] The O-H distance was constrained to 0.90 Å.
[[section]] The N-H distance was constrained to 0.88 Å.
2E.Steinwender, E.T.G.Lutz, J.H. van der Maas and J.A. Kanters, Vibrational Spectroscopy , 1993, 4, 217.
3 D.M.P.Mingos, T.E.Muller and D.J.Williams, J. Chem. Soc., Chem. Commun, 1994, ???.
4 H. S. Rzepa, M. L. Webb and D. J. Williams, J. Chem. Soc., Chem. Commun, 1991, 765.
5 H. S. Rzepa, B. J. Whitaker and M. J. Winter, J. Chem. Soc., Chem. Commun, 1994, 1907; see URL http://www.ch.ic.ac.uk/rzepa/RSC/CC/4_02963A.html; O. Casher, R. Chandramohan, M. Hargreaves, P. Murray-Rust, R.Sayle, H. S. Rzepa and B. J. Whitaker, J. Chem. Soc., Perkin Trans 2, 1995, 7. See also the URL http://www.ch.ic.ac.uk/rzepa/RSC/P2/4_05970K.html
6 F. H. Allen, J. E. Daview, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Comp. Sci., 1991, 31, 187.
7 J. L. Atwood, D. D. McNicol and J. E. Davies, Inclusion Compounds, Vol 2. Academic Press, 1984.
8 D. A. Jeffrey, J. M. Logan, W. M. Maier, J. Org. Chem., 1986, 51, 3206.
9 M.Parvez, S.K.Kurtz and I.Williams, Acta Cryst, 1990, C46, 165-166; H. W. Rauwald, K. Lohse and J. W. Bats, Angew. Chem. Int. Ed., 1989, 28, 1528.
10 G.S.D.King and L.Sengier-Roberts, J.Chem.Res., 1982, 22, 722
11 A. I. Gusev, D. Yu. Nesterov, A. F. Zhigach, R. A. Svitzin and E. S. Sobolev, Zh.Strukt.Khim., 1978, 19, 180.
12 J. D. Wallis and M. Pilkington, University of Kent, personal communication.
13 H. S. Rzepa, unpublished result.
14 A. D. Holmes and G. Pooley, University of Cambridge, personal communication. We are grateful to Dr Holmes for sending us the fractional coordinates for this system.
Figure Captions.
1. The molecular structure of 3.
2. The H-bonded cyclic tetrameric array formed by the crystallographically independent molecules of 3 in the crystal. The T-type aromatic-aromatic edge-to-face interactions are mediated by broken lines.
3.The centre-tetramer C-H...H interactions in the crystals of 3. The remaining three members of each peripheral tetramer have been omitted for clarity.
4. The molecular structure of 4.
5. The S6 symmetry H-bonded cyclic hexameric arrangement of the molecules of 4 in the crystal.
6. The cyclohexane-like chair conformation formed by the six H-bonded hydroxyl groups within each hexamer.
7. The molecular structure of 6 showing the intramolecular C-H...π interaction.
8. The pattern of disorder present in all of the crystals of 6 studied. Broken atoms and open bonds correspond to the minor occupancy orientation.
9. A centrosymmetrically related pair of molecules of 3 in the crystal showing the orientation of the amino N-H group in one molecule and the acetylenic bond in the other, and vice versa.
10. The molecular structure of 7.
11. Part of one of the H-bonded chains of molecules in the crystals of 7.