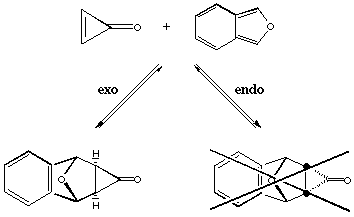Exo selectivity of the Diels-Alder addition of cyclopropenone and 1-3-diphenylisobenzofuran
- Introduction
Figure 1.1 : Diels Alder addition of cyclopropenone and 1-3-diphenylisobenzofuran
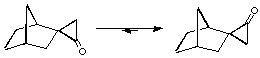
The Diels-Alder addition of cyclopropenone and 1-3-diphenylisobenzofuran yields only the exo adduct (Figure 1.1). This reaction was first made by Breslow and Oda[8]. In a recent publication[9], it was proposed that this exo preference is due to a thremodynamic effect. The authors, Cordes, de Gala and Berson, compare this exo-endo transformation to another rearrangement on the cyclopropanone shown on Figure 1.2. This conversion can be carried out at subambient temperature and the kinetic measures give an activation energy of 19 kCal/mol. They believe that the endo adduct is first synthetized and then rearranges immediately to give the exo product.
Figure 1.2 : Thermodynamic conversion of diastereomeric cyclopropanones
They refer to an intramolecular interaction between the carbon carbonyl and the bridging oxygen to explain the stabilization of the exo-isomer. This interaction looks like a Dunitz-Bürgi attack on a carbonyl. Could this interaction exist in the exo transition state? The fact that they can not prove easily the synthesis of the endo isomer suggests that the selectivity can be found earlier in the reaction process.
This project, then, has several aims :
- Analysis of the exo- and endo-isomer to verify the origin of the stabilization in the exo product
- Modelling of the transition states to measure the kinetic factors
- Comparison with similar reactions where the oxygen in the furan group has been replaced by other atoms.
- Experimental Results
Heat of Formation (kCal/mol)
PM3 / RHF |
| Exo Adduct | Endo Adduct
|
| 83.02 | 85.35 |
Table 1.1
It is obvious that the exo isomer is more stable. The distance between the carbonyl carbon and the bridging oxygen is 2.604 Å (PM3 value) / 2.54 Å (X-ray structure). We have a Van der Waals contact between these two atoms.The angle O...C=O is 124 ° (PM3 value) / 121 ° (X-ray structure). This is only 15° more than the Dunitz-Bürgi angle (105°) of the non-perpendicular attack on a carbonyl. We can say that there is a stabilization due to this interaction, that is not in the endo isomer.
Heat of Formation (kCal/mol)
PM3 / RHF |
| Exo Transition State | Endo Transition State
|
| 135.50 | 136.80 |
Table 1.2
When we refer to the Diels-Alder addition of two cyclopentadienes that exclusively yieldn the endo adduct, calculations give a exo transition state lower in energy than the endo by 0.8 kCal/mol. Furthermore, the difference here would appear to favour the exo transtion state. We can believe that there is a small kinetic exo preference but this is not so easy to prove.
Using the Woodward-Hoffmann theory of frontier orbitals to explain the endo-exo selectivity in Diels-Alder addition, we can find an HOMO-LUMO interaction* that will favorize the exo approach of the cyclopropenone.
Reaction on similar compounds
Calculations were carried out on the Diels-Alder addition of cyclopropenone to 1-3-diphenylbenzofuran derived compounds. The two phenyl groups were replaced by hydrogens.
We have been studying :
- benzonfuran ( -O-)
- benzocyclopentadiene (-CH2-)
- benzothiophene (-S-)
- benzopyrrole(-NH-)
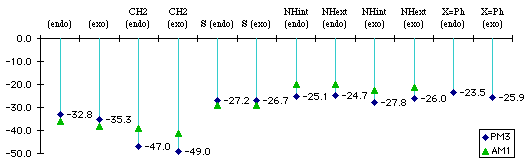
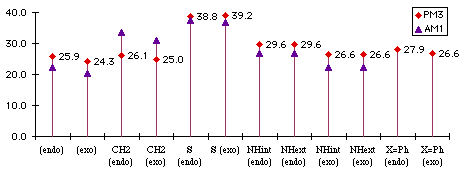
We make the distinction in benzopyrrole between isomers with the H pointing toward the benzene ring. This distinction may not necessarily be found in real compound, especially if there is a low energy-barrier between the two conformers. Both reaction enthalpies (Graph 1.1) and activation energies (Graph 1.2) were computed.
Graph 1.1 : Reactions Enthalpies (kCal/mol)
Replacing the phenyl (X=Ph) with hydrogen does not change the difference between endo and exo product. The addition with the phenyl substituted benzofuran is less exothermic because we lose conjugation between the two phenyls via the diene, whereas with the two hydrogens we do not.
The addition with the benzocyclopentadiene is the most exothermic because we do not lose any kind of partial aromaticity that can exist between the diene and the lone pairs of oxygen, nitrogen or sulfur.
The size of the sulfur atom destabilizes the exo product : there is a van der Waals contact between the sulfur and the carbonyl carbon. This explains exo and the endo products have almost the same energy.
Graph 1.2 : Activation Energies (kCal/mol)
The difference between diphenylbenzofuran and benzofuran is small. The two phenysl are still conjugated in the transition state. This confirms that we have early transition states : they looks more like the reactants than like products.
The addition on benzothiophene has the biggest activation energy, which is probably due to the size of the sulfur atom. Its van der Waals' radius is 1.85Å whereas that of oxygen is only 1.4Å and nitrogen 1.5Å. With the correction we have found necessary to make with cyclopentadienes, we can be sure that this reaction yields only the endo adduct, but this reaction will be the most difficult and will require high temperatures because it has the highest activation energy. For the same reason, we can think that the addition of the benzopyrrole will yield the exo product, but we ca not guess what the product of the addition of cyclopropenone to benzocyclopentadiene will be.
- Conclusion
The main characteristic of the Diels-Alder reaction that has been presented here is its exo selectivity. The mechanisms that are really involved in this selectivity are not well understood. We have tried to show how hypotheses such as thermodynamic rearrangements or HOMO-LUMO interactions can be studied and either rejected or confirmed. This can only be done within a model for the Diels-Alder reaction and its transition state.
Using Woodward and Hoffmann frontier orbitals, we can confirm the existence of a HOMO-LUMO interaction that could favour the exo-product. We should carry out ab initio calculations to confirm the fact that the activation energy for an endo attack is higher than for an exo attack. We can guess that the selectivity is due to kinetic factors.
This mini-project illustrates the use of several different modelling tools for the study of one reaction :
- Thermodynamics and kinetics : reaction enthalpies and activation energies.
- Geometric parameters.
- Molecular Orbital theory.
To prove the interest and the relevance of the models, we should attempt the reactions on benzocyclopentadiene, benzopyrrole and benzothiophene. The exo-endo selectivity would be a perfect test for the validation of the semi-empirical calculations.
*Exo selectivity of the Diels-Alder addition of cyclopropenone and 1-3-diphenylisobenzofuran
A molecular orbital approach (Figure 1.3)
In a semi-empirical calculation (MOPAC, PM3 parameters) on cyclopropenone, we have the following eigenvalues for the energy of each orbital (table 1.3):
| -14.3 eV | HOMO-3
|
| -11.1 eV | HOMO-2
|
| -10.2 eV | HOMO-1
|
| -0.1 eV | HOMO
|
| -1.3 eV | LUMO
|
Table 1.3
We have to take in account the HOMO of the double bond (HOMO-1). The HOMO (-0.1 eV) corresponds to the lone pair. Because the HOMO-2 is very close to the HOMO-1, it plays a important role in this frontier orbitals model.
The major difference between 1-3-diphenylisobenzofuran (1) and 1-3-diphenylisobenzopentadiene (1') can be found in the LUMO. Part of the LUMO of (1) is located on the oxygen. There is an interaction between the LUMO of the diene and the HOMO -2 of the dienophile ( which is the HOMO-1 of the double bond). The interaction is shown with the arrows in figure 1.3. This explains why exo addition is prefered with (1). This interaction does not exist with (1'). In both case there is a small interaction between the (HOMO-1) of the dienophile and the LUMO of the diene which suggests an endo product. In (1') this is the only selective interaction and we will have the endo product.
Steric effects could also be important and prevent the dienophile from approching the CH2 compound (1') in an exo position. These steric effects do not exist in (1).