Synthesis
Several total synthese of rapamycin have been reported3,4as well as many fragments and part-syntheses. Rapamycin is a complicated molecule comprising a 31-membered ring including a pipecolinyl group and pyranose ring, a conjugated triene system and a tri-carbonyl region. It also has 15 chiral centres, meaning the number of possible stereoisomers is enormous. The synthesis of rapamycin therefore presents a huge challenge to synthetic chemists.
In the following synthesis, published in three separate papers5,6,7two fragments of C10-C21 and C22-C42 are prepared separately, before being combined to give the total synthesis of rapamycin. Only the main outline of the synthesis will be shown as it is too long and complicated to show in great detail. For the full experimental details of the synthesis see the literature (ref. nos. given above).
In the retro-synthesis shown the molecule is disconnected at the ester group next to carbon 1 and the C21-C22 double bond of the triene to give the synthetic precursors 2 and 3. Further disconnections of 3 will be shown later. First the C10-C21 fragment is synthesised.
Synthesis of C10-C21 fragment
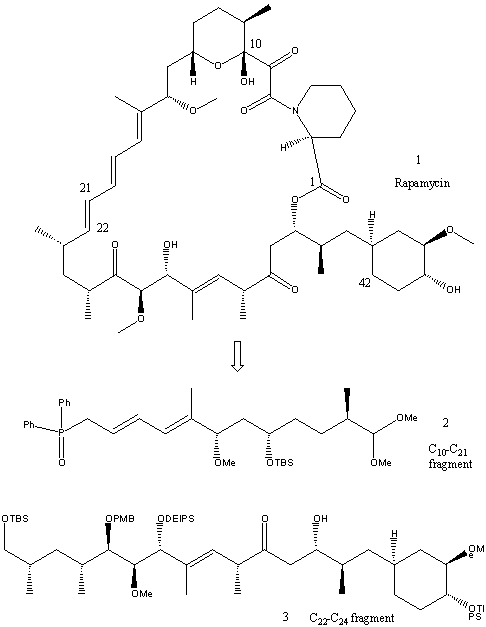
The synthesis uses (R)-methyl 3-hydroxy-2-methylpropionate (8) as a starting material.
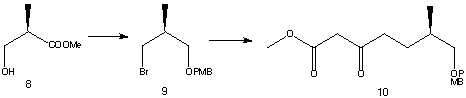
The starting material 8 is converted to an alcohol by a four-step process; protection of the alcohol as aTHP ether followed by reduction, ether formation and deprotection steps. Substitution of the hydroxyl group in the product for a bromine leads to the formation of the bromide 9. Reaction of 9 with methyl acetoacetate gave ester 10.
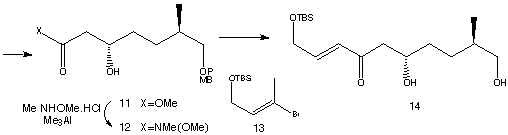
Catalytic reduction of 10 using the conditions of Noyori produced ester 11, which was then converted to its Weinreb amide 12. Overall, compound 12 was produced in 54% yield from an inexpensive starting material. Vinyl bromide 13 was metalated with t-BuLi and the resulting vinyllithium was combined with 12 and the PMB-protecting group was removed to give 14. The remaining carbonyl group in 14 was selectively reduced to a hyrdoxy group. In order to differentiate the 1,3-diol a lactol was formed, where one hydroxy group ended up in the ring. To acheive this an oxidation was performed using RuCl2(PPh3)3 resulting in formation of a lactol. The two remaining alcohol groups could then be methylated using MeI forming 15.
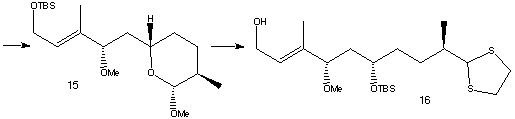
The lactol ring opening was achieved using TiCl4 and thiol HS(CH2)2SH to form a dithiolane. The freed alcohol was then protected as its TBS ether and the same protecting group selectively removed from the primary alcohol to form 16. To avoid removing the dithiolane group at a later stage in the synthesis the thio-acetal was converted to the dimethyl acetal 17 using PhI(OCOCF3)2 and methanol.
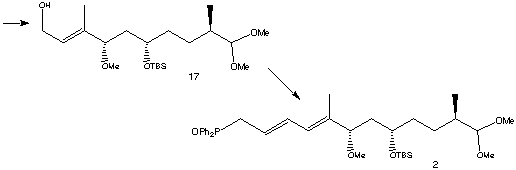
The next stage in the synthesis was to extend 17 for the building of the triene region. The terminal alcohol was oxidised to its aldehyde using BaMnO4 , then a Wittig reaction was carried out using Ph3P=CHCO2Et and CH2Cl2 to form the second double bond. Reduction of the ester group to an alcohol was carried out using DIBAL-H, then treatment with PPh3 and exposure to the air gave rapamycin fragment 2.
Synthesis of C22-C42 fragment
Here the retro-synthesis of 3 is shown, giving the three synthetic precursors 5, 6 and 7
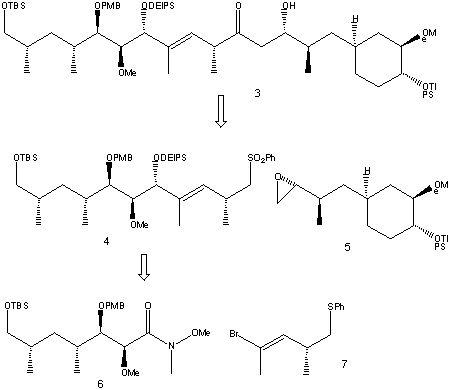
It was thought 4 could be obtained by alkylative coupling of a vinyllithium species generated from 7 to the Weinreb amide 6. The nucleophilic opening of epoxide 5 by the lithiated sulfone from phenyl sulfone 4 would then produce the desired fragment.
The ester 18 was used as a starting material to make fragment 6.
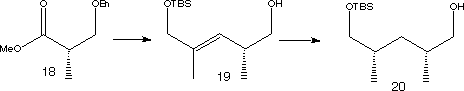
A Wittig reaction followed by reduction and protection steps produced 19. This was hydrogenated using a rhodium catalyst to give syn-dimethyl product 20. The minor anti diastereomer was successfully separated off. 20 was oxidised then underwent an aldol condensation to give adduct 21.
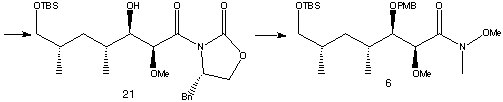
Transamination of 21 and protection of the alcohol with PMB resulted in amide 6, corresponding to the C22-C28 segment of rapamycin.
The vinyl bromide 7 was prepared using ester 22 as a starting material.
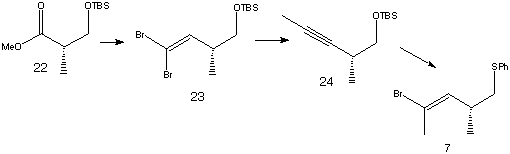
Reduction of 22 followed by dibromoolefination resulted in product 23. Acetylene 24 was prepared using n-BuLi, THF and MeI, then sulfenylation with Ph2S2 and bromination gave fragment 7.
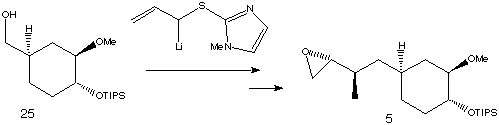
Iodination and alkylation of starting material 25 with the lithiated allylic sulfide shown followed by a number of further steps resulted in its conversion to fragment 5.
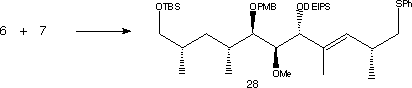
Fragments 7 was first converted to its vinyllithium using t-BuLi then combined with 6 forming an enone in 78% yield. Stereoselective reduction of the carbonyl group using Zn(BH4)2 gave an alcohol which was protected with DEIPS giving 28. The phenyl sulfide was oxidised to a sulfone using m-CPBA in excess pyridine.
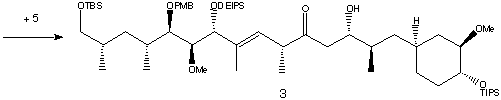
Lithiation and addition of the epoxide 5 resulted in the hydroxy sulfone in a 4:1 ratio of two diastereomers which were separated by HPLC. Metalation using n-BuLi followed by oxidation formed the total C22-C42 fragment.
Total synthesis of rapamycin through the combination of C10-C21 and C22-C42 fragments.
Fragment 3 (C22-C42) was treated with (S)-Boc-pipecolinal, followed by a Swern oxidation resulted in the aldehyde 29.
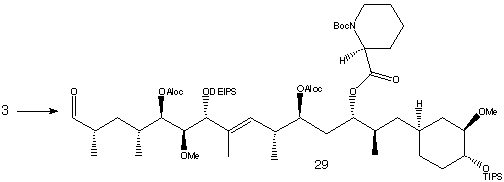
Condensation with the lithium salt of phosphine oxide 2 (C10-C21) produced the triene shown below.
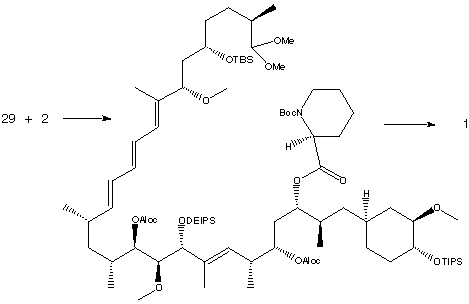
The triene was hydrolysed with pyridinium p-toluenesulfonic acid and an aldol reaction was performed. Treatment with triethylsilyl triflate produced an amino acid which was subjected to Mukaiyama macrocyclization conditions to form the 31-membered ring. Finally, deprotection steps were performed to give synthetic rapamyin (1). This was judged to be identical to natural rapamycin by comparison of physical properties, 1H-NMR, 13C-NMR, IR and UV spectral data.