|
|
|
|
In the past thirty years the fascination with cubane chemistry has continued unabated ever since the maiden construction of the cubane nucleus by Eaton and Cole. Since then, an impressive array of knowledge and understanding leading to the preparation of many different cubane derivatives has been amassed.
The reactivity of cubane and its derivatives can be illustrated by studying its reactivity and methods for synthesising specific target molecules. The reactions can be divided into the following categories:
![]()
Functional Group Transformations
It has already been mentioned that the carboxylic acid groups on cubane-1,4-dicarboxylic acid, like most other functional groups on cubane, are exceedingly well-behaved. Most typical functional functional group transformations can be applied successfully. This can be appreciated in the preparation of 1,4-dinitrocubane worked out at Chicago by Shankar et al in 1984. This is shown below:
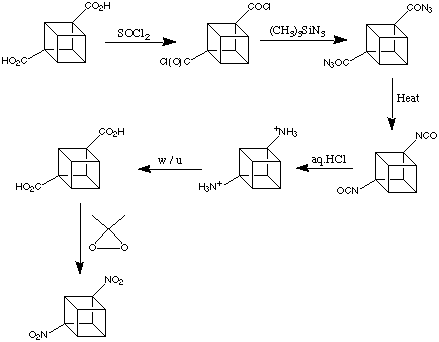
Here, modern versions of classical methodology are used. These conversions are as easy to do on the cubane system as on any standard carboxylic acid. Note, just as one example of the "convenient" behaviour of most cubane derivatives, that the intermediate cubyl-1,4-diisocyanate in the reaction scheme is a stable, crystalline material (m.p. 112 - 114 deg. C).
1,4-Dinitrocubane, the final product is perfectly easy to handle. It is less shock sensitive than TNT and remarkably stable thermally (decomposes at 260 degrees).
![]()
Substitution on the Cubane Framework
Functional group transformations are useful for preparing many mono- and disubstituted cubanes, as the corresponding carboxylic acids are readily available. But this does not suffice for the preparation of more highly substituted cubanes. As Reddy et al. have shown, substitution of the cubane framework by the way of the cubyl radical is fairly easy. However, as is usually the case with radical reactions, mixtures of products are obtained. Light induced iodination of cubane with tert -butyl hypoiodite gives mono-, di-, triiodocubanes, etc. The challenge is to achieve systematic and controlled substitution onto the cubane frame.
![]()
Amide activation for Ortho Metalation
The key idea here was to use neighbouring group activation to assist the metalation of cubane. In relation to this it was well known that an amide substituent on an arene aids lithiation in the ortho position by activation of the arene hydrogen atom.
There are certain similarities between cubanes and arenes: both have C-H bonds with enhanced s character, and in both the adjacent (ortho) substituents are forced to be coplanar. This is illustrated below:
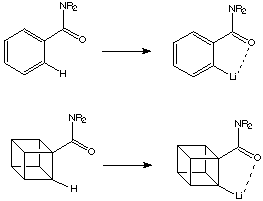
Thus it was speculated that it would be possible to lithiate cubanamides.
The seminal work was done by Graziano Castaldi in the Eaton Lab in 1985. He showed that a small equilibrium concentration of the ortho -lithiated derivative (centre, below) is indeed formed when the diisopropylamide of cubanecarboxylic acid is treated with a large excess of a strong, non-nucleophilic base like lithium tetramethylpiperidie (LiTMP). When the reaction mixture is quenched with CH3OD the recovered cubanamide contained approximately 3% deuterium in the 2 position (ortho); dideuteriated material could not be detected. This is shown below:
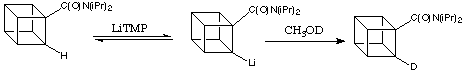
![]()
Transmetalation
In a key step forward, Castaldi showed transmetalation to be the golden step forward in the synthesis of substituted cubanes. When lithiation of a cubanamide is performed in the presence of mercury salts, the lithium compound first formed is quickly mercurated, and the very polar C-Li bond is replaced by the more nearly covalent (and relatively inert) C-Hg bond. In this way, the starting material in the scheme below can be completely converted into a metalated cubane.
![]()
The amide group is important in stabilising the intermediate lithiated cubane, but not the mercurated compound. Once lithium is replaced by mercury, the amide group is again able to assist removal of another ortho -hydrogen atom, and the process can continue.
Metalation / transmetalation is the basis of a complete synthetic methodology for the preparation of a great variety of substituted cubanes. Mercurated cubanes are useful for the formation of halocubanes, as the C-Hg bond is readily cleaved by halogens such as iodine.
![]()
Cubyl Grignard Reagents
Bashir-Hashimi at Geo-Centres introduced transmetalation with magnesium salts and thereby provided easy access to cubyl mono- and bis-Grignard reagents. The conversion proceeded by step wise lithiation / bromomagnesiation. The second metalation of the cubane framework occurs at a position remote from the first - quite understandable considering the polarities of the intermediates. Further metalation does not take place. In most cases, the overall conversion of these reactions is extraordinarily good; in the example shown below yields of 90-95% are easy to achieve in both small and large-scale runs.
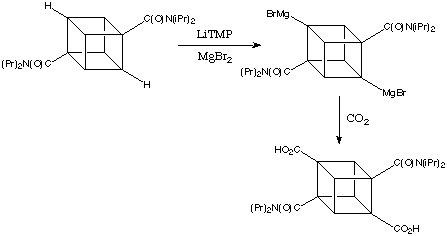
![]()
Ortho Magnesiation
In the course of extending to the synthesis of cubanes with different substitution patterns, the Eaton group found that treatment of the 2,4-dicyanocubaneamide with LiTMP gave two isomeric tetrasubstituted cubane carboxylic acids after carboxylation.
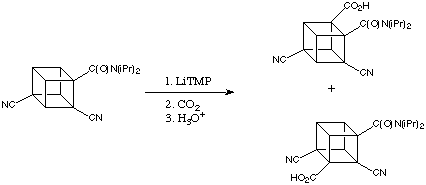
The electron withdrawing effects of the cyano groups along with the amide stabilises both intermediate lithiated cubanes so well that only a small excess of LiTMP is needed to achieve fairly complete deprotonation even at -78 degrees C. In the initial lithiation step, the activation of a cubane C-H bond by two flanking cyano groups rivals the ortho activation by the one amide group. This results in competitive lithiations and a mixture of products.
In attempts to achieve greater selectivity in this reaction, the effect of adding MgBr2 was examined. When the ratio of LiTMP to MgBr2 was 1.5:1, the product ratio was improved to 9:1 favouring carboxylation ortho to the amide function. This suggested an important and previously unknown reaction: direct magnesiation by TMPMgBr (a Hauser base) and / or by (TMP)2Mg. This idea has now been expanded into a powerful new process dubbed ortho magnesiation and it has been subsequentially found that magnesium amide bases are exceedingly useful in synthesis.
When dicyanocubanamide (below) is treated with TMPMgBr or (TMP)2Mg the only product after carboxylation (90% yield!) is that which contains the carboxyl group is next to the amide activating group.
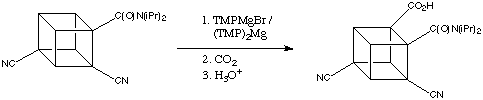
This reaction proceeds well even at -78 degrees C. The inductive effect of the cyano group clearly enhances the reaction, but the cyano group itself is not an ortho director for magnesiation.
![]()
Alternative substitution patterns
Although activation by a single amide group suffices for the metalation of cubanes, the presence of additional electron with drawing groups enhances reactivity enormously. 4,Cyanocubanamide is much more reactive than the unsubstituted amide. In cases when the electrophile can co-exist with the base it is possible to substitute all three positions ortho to the amide in a wonderfully simple, one-pot reaction proceeding by sequential metalations. Thus, for example, 4,cyanocubanamide can be converted directly into the tri(tert -butylcarbonyl) derivative, shown below.
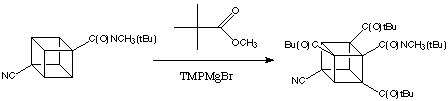
As the Baeyer-Villiger oxidation of tert -butyl cubyl ketones is completely specific - only the tert -butyl group migrates - triketone (below) can be converted easily to the polycarboxylated cubane:
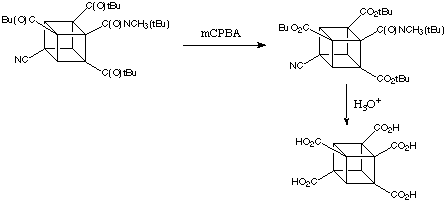
It is fair to say that almost any combination or permutation of functional groups and positions on the cubane nucleus can be achieved now by fairly simple chemistry.
The Eaton group have even synthesised cubanes with fused heterocyclic rings, an example of this is shown below.
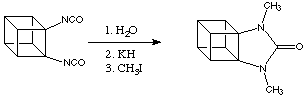
Quite different from expectation these "heteropropellano cubanes" are remarkably stable. The nitrogen atoms can be alkylated or nitrated without interferance from ring-opening reactions. Interestingly, the C-C bond shared by the urea ring and the cubane system is substantially shorter than the equivalent bond in cubanes or cyclic ureas. The origin of this effect is not known.
![]()
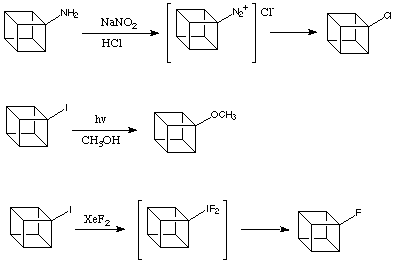
The cubyl cation
One of the biggest surprises in the research into cubane was the discovery that the cubyl cation is much more accessible than expected. Everything about the cubyl cation seems unfavourable:
Ab initio calculations (6-31G*, without electron correlation) place the cubyl cation about 20 kcal mol-1 higher in energy than tert -butyl cation and about 5 kcal mol-1 higher in energy than 1-norbornyl cation.
Even in the light of the above evidence there are numerous reactions that might involve the intermediacy of the cubyl cation.
![]()
|
|
|