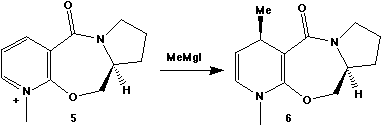Technique 8: Molecular Modelling.
- Objectives
-
Chemical Modelling Projects
-
Detailed Program System Instructions
- To become familiar with some common techniques for
molecular modelling, via "real" chemical examples
- To use molecular mechanics to predict the geometry and
regioselectivity of the hydrogenation of cyclopentadiene
dimer, the stereochemistry of nucleophilic addition of a
Grignard reagent and the conformation of a large ring ketone
intermediate to the anti-cancer drug Taxol, via a
choice of two programs (Chem3D) (< 3 hours)
- To use molecular mechanics to investigate Bredt's rule
for Bridgehead alkenes as a function of ring size using
Chem3D. To experience the skills involved in distinguishing
between local and global minima. (< 4 hours)
- To use semi-empirical molecular orbital theory to
investigate the regiospecificity of the electrophilic
functionalisation of a bicyclic diene using Chem3D (< 2
hours).
- To use ab initio molecular orbital theory to
investigate the frontier orbitals (and optionally the
transition states) involving reaction of a highly strained
bridgehead alkene, using an ab initio/density
functional programs (Gaussian03) (< 3 hours).
- To use density functional molecular orbital theory
to predict the 13C of your structure for your unknown
compound (< 4 hours)
- To perform searches of the literature in order to cite in
your final report any relevant references to each experiment
as appropriate.
Back to index
Introduction.
A general introduction to the Molecular Mechanics (MM) method
can be seen here. The present
techniques illustrate several more complex applications of this
method to typical chemical problems and the type of information
that such modelling is capable of providing. This involves
optimising molecular geometry to an energy minimum and
analysing the final energy in terms of bond length and angle
strain, steric effects and van der Waals contributions.
Before discussing specific applications of such a model, it is
worth noting some of the limitations of the molecular mechanics
approach. It is essentially a parametric method, using data
from experimentally well characterised and known molecules. It
is therefore used as an interpolative rather than an
extrapolative technique, which cannot stray too far from "known
chemistry". Thus it is not easily possible to model "kinetic
control" of a reaction using the standard approach, since that
requires knowledge of the transition state structure and
energy. For the same reason, new molecules with unusual bonding
are rarely amenable to modelling, and recourse has to be sought
in the full quantum mechanical treatment of the system.
Similarly, for molecular properties such as stereoelectronic
effects, aromaticity, hyperconjugation and frontier orbital
interactions which require a knowledge of the electron
distribution within the molecule, recourse has to be made to
quantum mechanical methods such as molecular orbital theory.
Finally, molecular mechanics parameters are available only for
certain types of bonds, and frequently are not available for
many functional groups. Metal ions are also a category less
easily handled at present by this type of model.
You will be using the Allinger MM2 or MM3 molecular
mechanics models1 together with Chem3D. MM2 and MM3
are particularly appropriate for hydrocarbons.
Chem3D produces an energy (in kcal mol-1) together
with optimised values for bond lengths, angles etc. This energy
is a rather odd quantity. It is NOT related to any
thermodynamic quantity such as ΔH, and
energies obtained using two different force fields CANNOT be
compared. You CAN however compare two energies calculated using
the same force field for two different ISOMERS. You can also
calculate energy differences for simple reactions such as the
hydrogenation of alkenes, particularly if this is compared
across a series of related reactions. The energy itself can be
dissected into contributions from the stretching (str),
bending (bnd), torsion (tor), van der Waals
(vdw) and hydrogen bonding (H-Bond) energy terms.
Each term indicates the deviation from "normality" of the
particular function. For example, a very positive stretch term
would indicate the predicted bonds are far from the "natural"
lengths, due to some geometrical feature of the molecule.
Comparing these terms across say two isomers provides a natural
explanation for why one isomer may be more stable than the
other.
Back to index
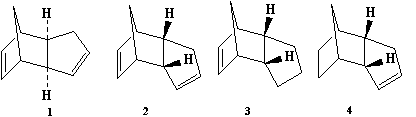
Cyclopentadiene dimerises to produce specifically the endo
dimer 2 rather than the exo dimer 1.
Hydrogenation of this dimer proceeds to give initially one of
the dihydro derivatives 3 or 4. Only after
prolonged hydrogenation is the tetrahydro derivative formed.
The modelling technique here involves calculation of the
geometries and energies of all four species 1-4.
The relative stabilities of the pairs of compounds
1/2 and 3/4 should indicate which
of each pair is the less strained and/or hindered in a
thermodynamic sense. The observed reactivity towards
cyclodimerisation and hydrogenation can of course be due to
either thermodynamic (ie product stability) or kinetic
(ie transition state stability) factors. In pericyclic
reactions in particular, regio and/or stereoselectivity is
controlled by the electronic properties of the molecules
(stereoelectronic control), and hence can only be understood in
terms of eg the molecular wavefunction (cf 2nd
year lectures on pericyclic reactions). On the basis of the
results obtained from the molecular mechanics technique you
should be able to suggest whether the cyclodimerisation of
cyclopentadiene and the hydrogenation of the dimer is
kinetically or thermodynamically controlled.
Procedure
Using Chem3D, define the two products 1 and 2 and
optimise their geometries using the MM2 force field option. In
the light of the above discussion, relate your results to the
observed mode of dimerisation. The two products of
hydrogenation 3 and 4 can be similarly compared
so that a thermodynamic prediction of the relative ease of
hydrogenation of each of the double bonds in 2 can be
obtained. Analyse the relative contributions from the
stretching (str), bending (bnd),torsion (tor), van der Waals
(vdw) and hydrogen bonding (H-Bond) energy terms in terms of
the relative stability of 3 and 4.
Back to index
The optically active derivative of prolinol (5) shown
below reacts with methyl magnesium iodide to alkylate the
pyridine ring in the 4-position, with the absolute
stereochemistry shown in 6.
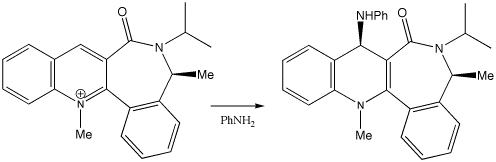
Another recent example of this type of stereocontrol is
shown below.
This reagent acts to transfer the NHPh group back to e.g.
esters etc in a reverse of the reaction by which it was formed.
On the basis of your previous calculations, can you speculate
on the origin of the stereocontrol in the formation of this
reagent? This reaction, by the way, links into the theme of
atropisomerism discussed in the section on Taxol below, as a
reagent for generating amides exhibiting (modest)
atropenantioselectivity (a phenomenon induced by steric
inhibition of rotation).
Use Chem3D to construct a model of the reactant, using the
template option to introduce the proline sub-structure (do NOT
include the MeMgI component. If you do try, what happens?).
Minimise the model using the MM2 force field (Chem3D offers no
option). Focus particularly on the geometry of the carbonyl
group, and its (quantitative) orientation with respect to the
aromatic ring. The "energies" of the molecules are much less
important in this context. Discuss the geometry obtained in
terms of the mechanism of reaction and in particular why the
methyl group is delivered with the stereochemistry shown above.
How can this simple model be improved (ie what is
missing from the present model?).
Reference:
- A. G. Shultz, L. Flood and J. P. Springer, J. Org.
Chemistry, 1986, 51, 838. DOI: http://dx.doi.org/10.1021/jo00356a016
- Leleu, Stephane; Papamicael, Cyril; Marsais, Francis;
Dupas, Georges; Levacher, Vincent. Tetrahedron:
Asymmetry, 2004, 15, 3919-3928. DOI: 10.1016/j.tetasy.2004.11.004
Back to index
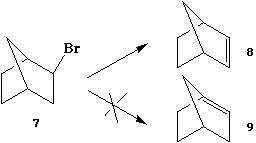
A number of years ago, Bredt noticed that in compounds
containing two or more fused rings systems, isomers containing
a double bond at the bridgehead position tended not to form.
Subsequently, a number of such alkenes have indeed been
prepared, and the double bond has been found to be highly
reactive3. Since the molecular mechanics approach is
capable of calculating ring strain quite accurately, it should
in principle be possible to formulate rules for the stability
and reactivity of bridgehead hydrocarbons based on such
calculations alone. Any rule must be formulated by comparing
several structures in a relative sense. There are at least two
possible approaches to doing this.
- One can calculate the heat of hydrogenation of a series
of bicyclic hydrocarbons as a function of ring size, and
compare the values obtained with eg cyclohexane as a
typical unstrained system.
- One can compare the calculated energies of the two double
bond isomers of the bicyclic hydrocarbon in which the double
bond is either at the bridgehead position or located away
from it.
Procedure
Devise a maximum of four bridgehead bicyclic alkenes
which vary in ring size in some systematic manner. Using the
Chem3D program and the MM2 force field, calculate the heat of
hydrogenation of all four compounds, and compare graphically
the results with eg the heat of hydrogenation of
cyclohexene. Is it possible to devise a hydrocarbon which is
more difficult (thermodynamically) to hydrogenate than
cyclohexene ? (Hint: see ref 2). If it is possible in your
examples to produce double bond isomers where the unsaturation
is not at the bridgehead, attempt to see if there is a
correlation between the isomerisation energy and the heat of
hydrogenation. Discuss your conclusions in the laboratory
report.
References.
1. Molecular Mechanics: E. Eliel and N. L. Allinger,
"Topics in Stereochemistry", Wiley, 1978.
2. W. F. Maier and P. von R. Schleyer, J. Am.
Chem. Soc., 1981, 103, 1891. DOI: 10.1021/ja00398a003
3. G. A. Kraus, Y.-S Hon, P. J. Thomas, S. Laramay,
S. Liras and J. Hanson, Chem. Rev., 1989, 1591. DOI: 10.1021/cr00097a013
4. I. Novak, J. Chem. Inf. Model., 2005,
45, 334-8. DOI: 10.1021/ci0497354.
Back to index
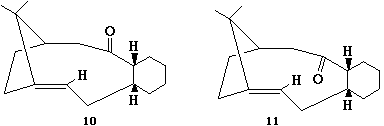
A key intermediate 10 or 11 in the total
synthesis of Taxol (an important drug in the treatment of
ovarian cancers) proposed by Paquette is initially synthesised
with the carbonyl group pointing either up or down. On
standing, the compound apparently isomerises to the alternative
carbonyl isomer. This is an example of atropisomerism".
Clearly the stereochemistry of carbonyl addition depends on
which isomer is the most stable. It is also noted that during
subsequent functionalisation of the alkene, this reacted
abnormally slowly!
Procedure
Using molecular mechanics (MM2) to determine the most stable
isomer 10 or 11, and to rationalise why the
alkene reacts slowly (hint: see previous exercise!). Pay
particular attention to the conformation of the resulting
optimised structure, to see if any aspect of this structure
could be improved by further minimisations (preceeded if
necessary by a manual edit of the structure to move atoms into
more correct orientations).
References:
- S. W. Elmore and L. Paquette, Tetrahedron Letters,
1991, 319; DOI: 10.1016/S0040-4039(00)92617-0
- See J. G. Vinter and H. M. R. Hoffman, J. Am. Chem.
Soc., 1974, 96, 5466 (DOI: 10.1021/ja00824a025) and 95,
3051 for another nice example of atropisomerism.
- Another well known example is within Vancomycin: J.
Am. Chem. Soc., 1999, 121, 3226. DOI: 10.1021/ja990189i
- An interesting variation is of "atropenantioselective
cycloetherification" (G.ÊIslas-Gonzalez,
M.ÊBois-Choussy and J.ÊZhu, Org. Biomol.
Chem., 2003, 30-32. DOI: 10.1039/b208905. See also Leleu,
Stephane; Papamicael, Cyril; Marsais, Francis; Dupas,
Georges; Levacher, Vincent. Tetrahedron:
Asymmetry, 2004, 15, 3919-3928. DOI: 10.1016/j.tetasy.2004.11.004
Back to index
Introduction.
In part 1, the strengths and weaknesses of a purely mechanical
molecular model were illustrated. In particular, the
endo stereoselectivity in Diels Alder cycloadditions was
attributed to "secondary orbital" interactions, which the
Molecular Mechanics approach cannot handle. In this section,
several such electronic aspects of reactivity will be
illustrated, showing how explicit consideration of the
electrons in molecules must be taken into account.
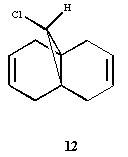
Orbital control of reactivity is illustrated in the reaction of
compound 12 with electrophilic reagents such as
dichlorocarbene or peracid;
In modelling such a reaction, we require a program where the
geometry of 12 can be predicted, and the energy of the
orbitals calculated and their form displayed graphically. Use
the Chem3D program and select the PM3 valence-electron
self-consistent-field MO method to provide an accurate
representation of the valence-electron molecular wavefunction,
and in particular of the HOMO (Highest Occupied Molecular
Orbital), presumed to be the most reactive towards
electrophilic attack.
Back to index
Using the Chem3D program, draw the molecule, and select
Calculations/MOPAC/Minimise energy option from the top menus,
and from the Theory pane, select PM3. [It might be advantageous
for you to process the drawn molecule first using Mechanics to
tidy it up a bit, and only then to subject it to MOPAC]. When
this is complete, select Gaussian/Compute Properties and Molecular Surfaces.
When complete (about 4 minutes), select View/ Molecular
orbitals. The HOMO appears by default. Select View/Select molecular orbital
to view the
HOMO-1, the LUMO, LUMO+1 and LUMO+2. Copy (as
bitmap) and paste each orbital into Word.
References.
1. T. L Gilchrist and R. C. Storr, Organic Reactions
and Orbital Symmetry, CUP, 1979.
2. B. Halton, R. Boese and H. S. Rzepa., J. Chem.
Soc., Perkin Trans 2, 1992, 447. DOI: 10.1039/P29920000447
Back to index
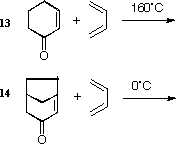
The different reactivity of 13 and 14 can also be
explained by the energies of the frontier orbitals, or more
accurately by the barriers to the transition states for the
reactions.
The energy of the HOMO will be calculated for both systems.
The one with the highest energy HOMO (least negative) is the
one likely to react fastest. In this case, you will calculate
the HOMO energies of 13 and 14 using Gaussian, which allow "ab initio"
wavefunctions to be calculated and displayed (the calculation is performed
without the butadiene, which is common to both and hence is neglected).
Procedures.
You can use the Chem3D, Gaussview programs or the brand new
SCAN to run a Gaussian calculation. Select
Gaussian/Minimise Energy from the top level menu, and in the
Theory section, specify Method: DFT=B3LYP (Density functional
theory), Wavefunction= Restricted Closed Shell and 6-31G Basis
set with Polarisation set to d. (or if you are in a
rush, use the much faster 3-21G basis set). HINT 1:
Pre-optimise the structure using a fast method such as
Molecular mechanics, before submitting the ab initio
calculation. If you do not do this, the latter will take much
longer! HINT2: Inspect your geometry for 14 carefully;
it is possible to get an unexpected (high energy) stereoisomer. Think about it!
Using the SCAN. The SCAN is: SuperComputer-At-Night.
With this, you can run larger molecules, or smaller molecules
at a much higher level of theory. Proceed as follows:
- Using the Chem3D or Gaussview programs to create a Gaussian
input file (a .gjf file).
- Go to https://scanweb.cc.imperial.ac.uk/uportal2/
and log in.
- Select Projects and create a project name suitable
for your needs.
- Select New Job, Chemistry Condor Pool, your
project, the.gjf file created earlier, and click on submit.
The Job will run overnight and can be collected from this web
page. In particular, if you select the Formatted
checkpoint file from output list, and download it,
Gaussview will open it and display the result of your
calculation. You can also open this file with Chem3D.
- The SCAN is powerful enough that if you wished, all the
molecules in this technique could be submitted using the
Gaussian program. You can submit multiple jobs, one after
another using this technique. You could also increase the
level of theory. In this case, change the basis set from
6-31G(d) to e.g. cc-pVTZ, or you could e.g. Include
vibrational analysis, which in fact will result in an
entropy correction to the energy, to give in effect a
ΔG for your energy.
Reference
- H. O. House, J. L. Haack, W. C. McDaniel, and D.
VanDerveer, Enones with strained double bonds. 8. The
bicyclo[3.2.1]octane system, J. Org. Chem., 1983, 1643-1654. DOI: 10.1021/jo00158a014
Back to index
The 13C spectrum of an organic molecule can be
predicted using two quite different methods.
- The first is a rule-based approach derived from a fragment library;
the MestreNova program uses this method. The advantage is that the prediction
is extremely rapid, and fairly general. The downside is that the accuracy
is only around 3-5 ppm, and does not take into account local conformations,
differential solvation of different groups, etc etc. If you want to try this
approach, some concise instructions are to be found here.
- The second is the so-called GIAO approach using quantum mechanical density
functional theory. The background to this, and a famous recent
example can be found in the article by Rychnovsky1
on a revision of the structure of Hexacyclinol. He
reports that the mean error for the 23 carbon shifts in the
predicted structure was around ± 1.8 ppm, with a maximum
error of around 5.8 ppm. An improved procedure which reduces
the mean and maximum errors by one half will be used
here2, although a number of caveats for successful
prediction should be noted. The most serious is that the method
is highly sensitive to the conformation of the molecule.
If various different conformations are possible (and for some
molecules, 100s of reasonable conformations can sometimes be
imagined), they should all be scanned by this method. Since
this is clearly not feasible in a reasonable time, you should
not expect a fit to your unknown compound to be as successful
as the preceeding errors might suggest.
- Important: Experiment 8 is designed teach how how to evaluate
an unknown compound by predicted its NMR chemical shifts. Your unknown
compound is a useful example to use, but you will not be graded here on whether
you have correctly identified it or not. If you do not yet know the identity of
the molecule, then attempt a reasonable guess. If that guess is not correct, you
will NOT be penalised at all during the marking of expt 8.
Procedure
You will need to sketch your molecule in ChemDraw Pro/Chem3D and perform
an initial refinement of its 3D geometry. If it contains only
simple elements (CHNO, Si, P, S, halogens) then the chances are
that a molecular mechanics refinement will be possible. At this
stage, whilst the calculations still take only a few seconds,
you might wish to investigate several conformational
possibilities to see which might be the lowest (but don't try
more than say 5). Some conformations can be preset (a
worthwhile one is to always try to get 6-membered rings into a
chair, and e.g. esters R-CO-O-R' oriented such that the R-C
bond is antiperiplanar to the O-R' bond). If the mechanics
procedure fails because of lack of parameters, try eg the
MOPAC/AM1 approach instead. If both of these fail, try the
Gaussian procedure, using the HF (Hartree-Fock) method and an
STO-3G basis set. This initial geometry will then have to be
refined/optimized using the following method.
- In Chem3D, go to Calculations/Gaussian/Create Input
File.
- Select Job Type/Minimise; Method DFT=mpw1pw91
- The Basis set
to be set to 6-31G(d,p)
- Save the resulting file to your H: drive, making sure it is saved as a Gaussian Input file, with the suffix .gjf.
- Find the file in Windows Explorer, and with a
right-mouse-click, open it with the WordPad program.
-
Delete all lines at the top, leaving only the following line, which
should be edited to show:
# mpw1pw91/6-31(d,p) opt(maxcycles=15) scf(conver=7)
NMR calculation for unknown compound
0 1
C 0 -4.35079359 2.69494079 -2.13433575
... ... ...
This shows the keyword line at the top, a blank line, a title card, another blank line, a
charge/spin card (we will assume that your unknown is
neutral and a singlet spin state) and the first line of atom
coordinates. Whilst you are at it, check to see if your coordinates have any
atom type designated Lp. If any such lines are present, delete the entire
line. Lp is a Lone-pair, and is sometimes added by the Molecular Mechanics
part of the program. However, if Gaussian sees it, it gets very confused, and
will not run at all!.
- Re-save this file, making sure you save it as TEXT and NOT RTF and that it retains the suffix .gjf.
- Go to https://scanweb.cc.imperial.ac.uk/uportal2/, log in,
and first create a project (it could simply be called unknown
compound). Then, New Job/Chemistry Condor Pool, then select
Gaussian/Your project, and finally the name of the Gaussian
input file you have just saved, along with a descriptive
title.
- You can view your job list, when a display of the type
shown below should appear:
Jobs in the pool tend to run overnight, and you will
probably have to wait till the next day for the job to
complete. The status of the pool can be inspected by
selecting Pools from the menu on the left:
If your suspected molecule is large (more than about 22
non-hydrogen atoms) it may require more powerful computer
facilities. If the job returns no output overnight, it may
well have run out of time (it has about 9 hours in which to complete
the calculation). You could run on a faster computer; contact Prof Rzepa in this case.
- When the job shows as Finished, select the Gaussian
Checkpoint file as the required output:
and download it (probably to the desktop, or wherever the browser tells you). Double-click the
file to open Gaussview (it may happen automatically)
and check that the optimised geometry
is still reasonable. Invoke File/Save as and replace
the original Gaussian input file you created with Chem3D. It
now has a fully optimised geometry, rather than the initial
sketch of before.
- If the system responds that the formatted checkpoint file does not exist
its quite probable that the calculation failed. Two common reasons for the failure are
- There was an error in the input .gjf file. A common error is the positioning or omission
of blank lines. Check with the above to ensure they are correctly positioned. Another
error is that the keywords are mis-typed.
- The calculation may have run for 9 hours and then run out of time (see above)
-
Repeat the Wordpad editing procedure as described above,
but this time ensure the top line contains the following
# mpw1pw91/6-31(d,p) NMR scrf(cpcm,solvent=chloroform)
NMR calculation for unknown compound
0 1
Br 0 -4.37079359 2.59494079 -2.03433575
- Resubmit this new input file for calculation as described
above. This will take less time to calculate than
before.
- When this second calculation is finished, download this
time the Gaussian Log file (instead of the checkpoint file).
Open this in Gaussview and from that program, select
Results/NMR (if the NMR keyword is greyed out, it means the
calculations was not in fact successful).
- From the Spectral display that appears, select the
C nucleus, and the appropriate Reference Value:
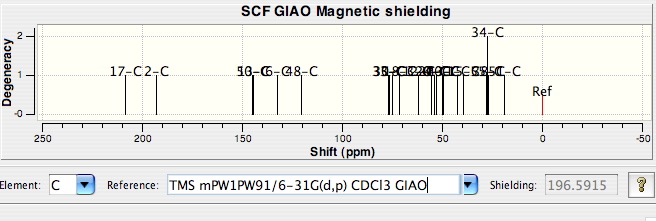
click on any peak to find out what its chemical shift is,
and compare with the spectrum that you have been issued
with.
- You should note that carbons attached to "heavy" elements
(particularly eg halogens) have shifts which need correction
for so-called Spin-orbit coupling errors. Typically, C-Cl needs correcting
by -3 ppm, C-Br
by -12 ppm, and C-I by about -28 ppm. Other
elements to be determined!2. Another systematic error present is
that the carbonyl of esters, amides etc tends to be out by about 5ppm. Use the
following simple correction for such carbons only:
δcorr = 0.96δcalc + 12.2.
- You can probably use your calculation to actually assign the 13C
shifts to the carbons of your molecule. If you spot one or more carbons out by
more than about 5ppm, its quite likely that you have the wrong conformation
of your molecule in that region (i.e. the method can actually be used for
conformational analysis).
- The method should work for other nuclei (except hydrogen, which requires
much greater accuracies to be really useful).
- Complete this section by returning to https://scanweb.cc.imperial.ac.uk/uportal2/ and click on the
publish link next to the job that carries the NMR prediction. This will deposit your calculation
into a so-called Digital repository, from whence it can be retrieved by anyone wishing
to find it. In this instance, it could be checked during the process of grading your final
report.
References
- S. D. Rychnovsky, Org. Lett.,, 2006,
13, 2895-2898. DOI: 10.1021/ol0611346
- C. Braddock and H. S. Rzepa, J. Nat. Prod.,
2007, submitted for publication.
The above technique is reliable for 13 shifts, but less so for
1H shifts. However, three-bond couplings can be predicted reasonably well
using a very rapid and simple method. To to this, you will need to have a 3D
model of your unknown, which should emerge out of your 13C prediction
in the preceeding section. The start point should be to use Chem3D to save
an MDL Molfile of your final coordinates. This can then be read into Janocchio,
to provide the coupling constants.
Back to index
In your report you should discuss your evaluation of each of
the techniques you use here. You should include at least three
literature references in addition to the ones given here.
The simplest way of producing a report of these experiments
is to select and copy diagrams from Chem3D
software to an Office 2003/2007 document. Please save as a .doc document
rather than the default .docx format.
Submit by email to org-8@ic.ac.uk.
Important: We have had, in the past, instances of
students copying another's molecule and orbital diagrams and
submitting these in their report. This is considered
plagiarism, and if detected will result in disciplinary
proceedings. If your diagrams for some reason are lost due to
computer failure, please discuss this with the
demonstrator.
Copyright (c) H. S. Rzepa and Imperial College London Chemistry
Department, 1994-2007.