The CAChe software can be run on the eMac Computers. There
are three licenses for such systems, two of which are fitted to
the eMac computers in the Perkin lab, and one is installed on
the eMac computer in the main computer room. Although you can
log into these machines using your conventional account,
unresolved issues (with file permissions) mean the CACHe
software does not yet run properly. As a temporary solution,
please log in as user cache and password
expt9.
At the end of this part of the experiment, you will probably
need to transfer files from these systems to a Windows system.
To do so, select Go/Connect to Server from the "Finder" window,
and enter afp://chnts1.ch.ic.ac.uk/mac-PC. Enter your
normal login and password, whereupon a volume will appear on
your desktop called Mac-PC. Save your files there (in your own
folder) and you can read them again using a Windows
computer.
-
To invoke the CAChe structure "Editor", find this in the
"Dock" that appears on the bottom of the Screen.
A new "Window" opens called Untitled - 0 and several new
menu items appear on the top of the screen. Select the atom
tool;
If there are appropriate templates, you could instead
start with one of those;
The default atom using the atom tool is sp3
hybridised carbon. Draw a carbon skeleton as follows;
- Click once on the drawing area, when a carbon atom
appears.
- Locate the cursor over the top of this atom, press the
mouse button, and KEEP IT PRESSED. Move ("drag") the mouse
while you are doing this to a new position and release the
button. A second carbon appears with a bond connecting it to
the first.
- If you "locate, press, drag and release" between two
existing atoms, the single bonds can be changed to a double
bonds.
-
To change carbon to a heteroatom, go to the atom menu,
press the mouse button, "drag" to the element you want and
release to select as the drawing tool;
Position the cursor on the atom you want to change, hold
down the apple key and click the mouse button.
-
You now have a structure with no hydrogens and the
incorrect configurations (ie sp3 etc) for the
atoms. The structure can be 'Beautified'
- You now have a complete structure which should fill the
window. To view this molecule from different angles, the
virtual "trackball" can be used.
- To rotate your view of objects on the screen, press the
Apple key and the SHIFT key at the same time and drag the
view with the mouse
- To translate your view of objects on the screen, press
the Apple key and the OPTION or ALT key at the same time and
drag the view with the mouse.
- To zoom and pan your view of objects on the screen, press
the Apple, OPTION/ALT and the CONTROL keyss at the same time
and drag the view with the mouse
- To scale objects, press Apple and CONTROL at the same
time.
- If you want to delete superfluous atoms, first chose the
select tool;
Select them by positioning the cursor on the first atom and
click the mouse. Now hold the SHIFT key down and keep it down
while you click on all the remaining atoms you want to delete.
Each will appear highlighted. Press the "Delete" key (often
shown with a reverse arrow) on the keyboard to delete these
atoms. If you make a mistake, undo from the Edit
menu will do just that. Whilst atoms are selected, you can
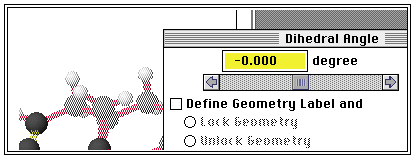
measure distances angles etc between them. Go to the Adjust
menu and select e.g. "Dihedral Angle" if you have four
atoms selected;
The display will tell you what the current angle is (if you
select only two atoms, you can measure a distance, etc).If you
type 0 from the keyboard, you will change the current value to
0deg. for the angle. Click on "Apply" and then "Done" to
implement the new value.
- The next stage is to optimise the geometry. Normally,
this would be performed first using Molecular Mechanics but
for a molecule as small as this one can use the program MOPAC
directly, as selected from Applications menu;
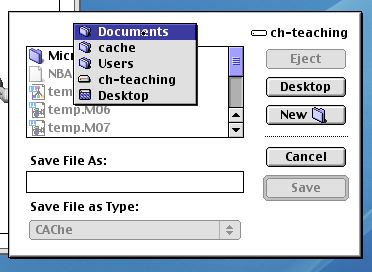
You will be first asked if you want to save your file to disk.
Please take care to save the file correctly. It should
be saved to the local machine hard drive, ideally in the path
Users/cache/Documents, and in this area you should create a New
directory for your experiment. Otherwise, there is a
possibility that you may overwrite another persons files [this
rather awkward requirement is necessitated because the CAChe
software does not work correctly when so-called network logins,
ie using your own private account, are used. Hence the need to
use the communal cache account for this experiment]. After
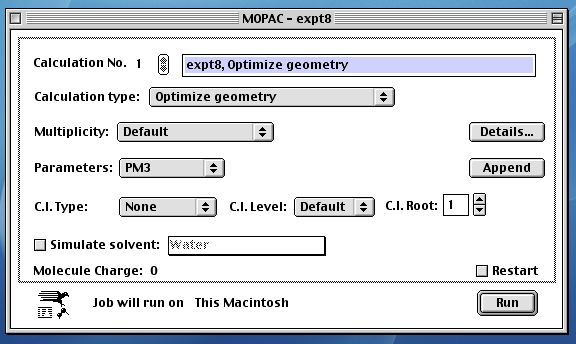
selecting OK, you enter the MOPAC module. All the default
parameters should have the following values. If they do not,
then reset them to the values shown below;
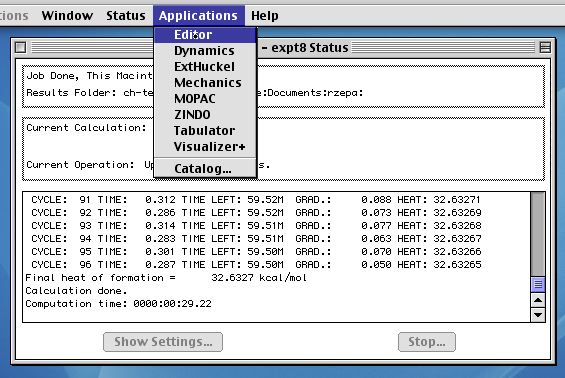
At this point, MOPAC will start running; the output looks
similar to that below. It may take 5 or so minutes to finish.
Using the PM3 parameter set, you should get an answer similar
to that shown below. If its significantly ( ie 0.5 kcal/mol)
different, you probably have a wrongly connected structure, or
an entirely different molecule to the requisite. Check your
structure again by return to the Editor;
You should inspect the final optimised structure at this
stage, not only for any anomalies, but also for any expected
(or unexpected) structural features. If you have time, you
could repeat with a different parameter set (PM5 is the latest,
and may even be better than PM3) and also establish if any
unusual structural features persist across different
semi-empirical MOPAC methods.
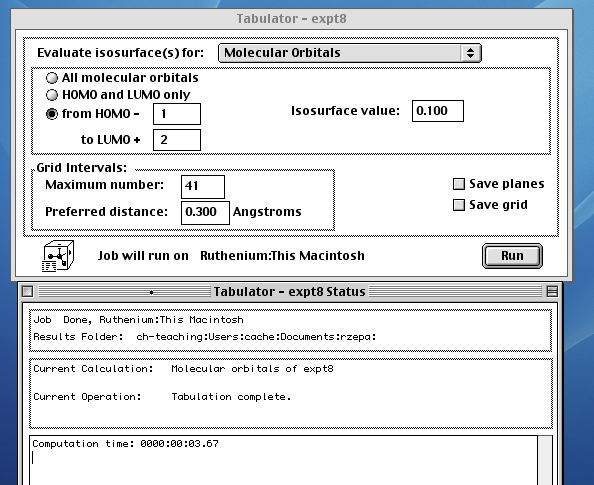
Next comes the Tabulator (from the applications menu), where
the required molecular orbitals will be evaluated. Choose only
those of interest;
If you have time, you could also compute the molecular
electrostatic potential at this stage (it takes about 20
minutes to produce).
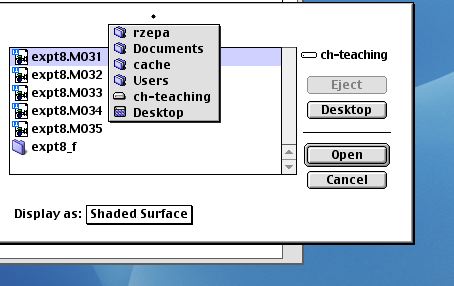
Next comes Visualizer+; you will have to select the orbital
you want by opening a surface from Visualiser.
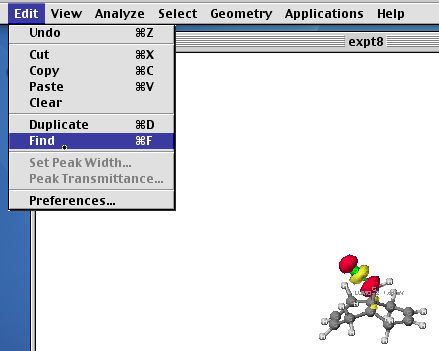
Resize the molecule if necessary using the Find command.
You should inspect each orbital in turn. From the nature of the
HOMO, can you predict the regiospecificity of molecule
12? In the literature reference, an argument is
presented for why the two π HOMO orbitals are different in
energy. Can you reproduce this argument by illustrating the
relevant orbitals? If you want to inspect the eigenvalues
(energy levels) or eigenvalues (coefficients) of the orbitals,
proceed as follows. ( The HOMO
should always have a strongly negative energy, e.g. -8 or more
eV. Note also 1 Hartree= 27.2 eV (you may need this conversion
factor if using other programs such as Gaussian).
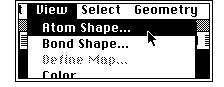
- Next you can display the calculated atomic charges. From
the View menu;
followed by;
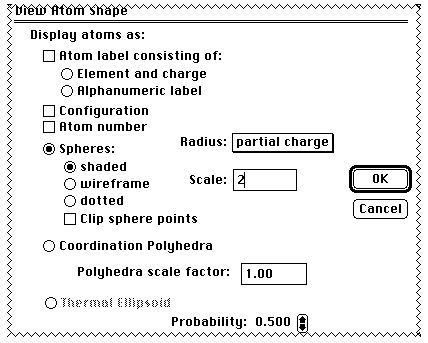
The scale should be set to 2.0 (or greater) from the default
value of 0.25. The size of the atom spheres now indicates the
charge on the atom (yellow negative, red positive). Comment on
these values in relation to the regioselectivity towards an
electrophile!
- You can copy the orbital window to the clipboard, and
paste the contents to a Word processor document to produce a
report of your experiment.
- To exit from Visualiser (or any of the other
programs);
Back to index