COMPUTATIONAL METHOD.
- Macromodel/Batchmin package [2] was used to perform a Molecular
Mechanic computational study on model cryptands 1c and 2c.
- a Monte Carlo Multiple-Minimum (MCMM) search [3] using the AMBER
united-atoms force field [2,4], and saving all conformations within 12
kcal/mol from the global minimum. After addition of hydrogens and lone
pairs, the structures were re-minimized using in turn the all-atoms
version of AMBER [2,4,5], and MM2 [2,6].
- all the found conformations were sampled several times, thus
ensuring convergence of the conformational analysis.
- MM2 proved to be the best method at calculating both reliable
structures and reliable energies.
- diastereoisomeric ratios were calculated from the Boltzmann
distrubution of all conformations within 2.5 kcal/mol from each global
minimum at T = 353K.
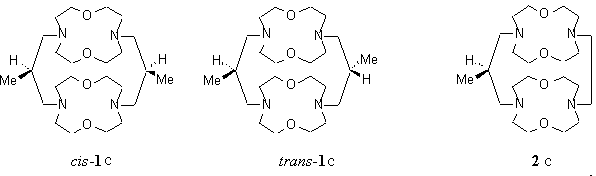
Amber u.a. : cis-1c / trans-1c = 97
/ 3
Amber a.a. : cis-1c / trans-1c
= 33 / 67
MM2 : cis-1c / trans-1c
= 78 / 22
Experimental : cis-1c / trans-
1c = 67 / 33