Gregory H. P. Roosa and Sundari Balasubramaniamb
a Chemistry Department, College of Science, Sultan Qaboos
University, P O Box 36, Al-Khod, Sultanate of Oman 123.
b Chemistry Department, Murdoch University, South Street, Murdoch
6150, Western Australia.
The utility of homochiral imidazolidin-2-ones in synthesis has only recently
been reviewed for the first time.1 Members of this class of readily
accessible chiral adjuncts have now proven themselves as competitive control
elements in both stoichiometric diastereoselective processes as well as in the
role of ligands for catalytic asymmetric synthesis.
This contribution describes some ongoing extensions in the use of the
ephedrine-derived (4R,5S)-imidazolidin-2-one auxiliaries 1
and 2 in further highly stereoselective coupling reactions.
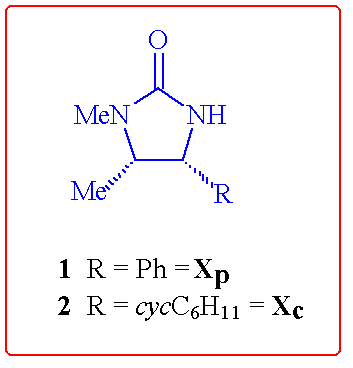
This current work has followed 2 distinct directions:
A. The stereoselective entry to [alpha]-amino phosphonates.
B. The stereoselective synthesis of [beta]-amino acids.
In a recent publication, Chung and Kang have reported an investigation of a
range of chiral auxiliaries to control the stereoselectivity of phosphite
addition to imines.2 Unfortunately, greatest control was achieved
with the relatively labour-intensive Oppolzer camphorsulfonamide
auxiliary.3 As depicted in Scheme 1, this one-pot procedure
may be performed in a highly stereoselective manner with the corresponding urea
derivatives of auxiliaries 1 and 2. As might be anticipated, the
poorest diastereomeric excess was obtained in the low sterically demanding case
of acetaldehyde (leading to 5c) where facial discrimination in the
intermediate N-acylimine is most challenging.
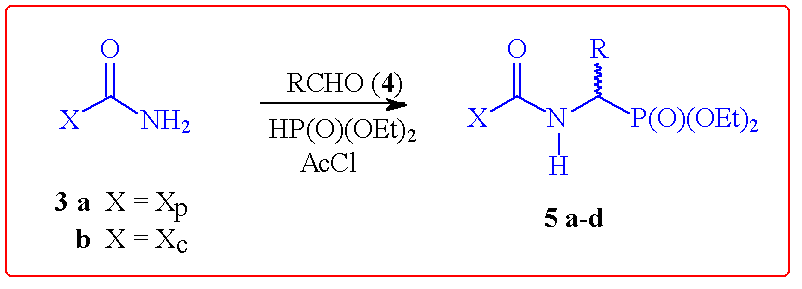
Scheme 1 Stereoselective [alpha]-Amino Phosphonate Formation
Urea
|
R
|
Yield
5 (%)a
|
d.r.%b
|
[[alpha]]D
|
3a
|
Ph
|
5a
(70)
|
>100:1
|
-48.4
|
3b
|
Ph
|
5b
(52)
|
>100:1
|
-1.2
|
3a
|
Me
|
5c
(42)
|
66:34
|
-6.4
|
3a
|
p-NO2Ph
|
5d
(68)
|
>100:1
|
-19.1
|
a Isolated yield after recrystallization. b As
determined by HPLC on a silica column
The correlation of the absolute sense of stereochemistry of the
[alpha]-aminophosphonates was provided by hydrolysis of 5a to give the
corresponding phosphophenylglycine. This product gave a rotation that is in
agreement with reported values for the (S)-configuration, and is consistent
with frontside attack of the phosphorous nucleophile on the s-cis/E
conformation of the N-acylimine intermediate.
One of the three primary strategies to [beta]-amino acids has been the addition
of enolate equivalents to imines.4 In an example of this approach, a
recent contribution by Wyatt and co-workers has sought to exploit the lithium
and titanium enolates of an Evans N-acyloxazolidin-2-one in this
role.5 This prompts us to report our findings in this area. Here,
whilst use of the ephedrine-derived auxiliary 1 again improved
stereoselectivity, the complete preference for syn selectivity is in
contrast to the reported work. The results in Scheme 2 are only for the
enolate based on auxiliary 1, since the alternative cyclohexyl analogue
proved to be extremely unreactive, possibly due to severe steric crowding
during approach of the Ti-complexed imine to the Ti-chelated enolate.
Scheme 2 Stereoselective Imine Coupling
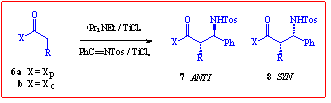
R
|
Ratio
7 : 8
|
7
Yield%
|
8
Yield%
|
Me
|
1:7.3
|
72
|
9
|
Ph
|
1:8.6
|
75
|
-a
|
a Not isolated
As can be seen, the reactions produced only 2 of the possible 4 stereoisomers
(as judged by 300MHz NMR) with good selectively between the minor anti
and major syn diastereomers. The high degree of crystallinity of these
high-melting products allowed for their ready separation by crystallisation.
The absolute stereochemistry of the products was confirmed by hydrolytic
removal of the auxiliary and comparison of the rotation of the N-Tos
amino acids with reported values.
REFERENCES:
1. Roos, G. H. P., S. Afr. J. Chem., 1998, 51, 1
2. Chung, S-K., Kang, D-H., Tetrahedron: Asymmetry., 1996,
7, 21.
3. Oppolzer, W., Chapius, C., Bernardinelli, G., Tetrahedron Lett.,
1984, 25, 5885.
4. Hart, D. J., Ha, D-C., Chem. Rev., 1989, 89, 1447.
5. Abrahams, I., Motevalli, M., Robinson, A. J., Wyatt, P. B.,
Tetrahedron, 1994, 50, 12755.
6. Drewes, S. E., Malissar, D. G., Roos, G. H. P., Chem. Ber.,
1993, 126, 2663.
SUMMARY EXPERIMENTAL
The imidazolidin-2-one auxiliaries 1 and 2, as well as
derivatives 6 were prepared as previously described.6
Preparation of Ureas (3): A stirred solution of the appropriate
imidazolidin-2-one (1 equiv.) in dry THF was treated with n-BuLi at
0°C. After 30 min, ethyl chloroformate (1 equiv.) was added and the
mixture stirred for 1 hr at 0°C before quenching with saturated
NaHCO3. The THF was removed under reduced pressure and the residue
partitioned between H2O and CH2Cl2. The
organic phase was dried (MgSO4) and concentrated to give the crude
ethyl formate derivative. This intermediate was taken up in further THF and
treated with excess concentrated aqueous NH3 (~25 equiv) and the
mixture stirred at room temperature overnight. The white precipitated product
was filtered, washed with H2O, and recrystallized from EtOAc.
(3a 95% yield, m.p.240°C, [[alpha]]D -2.3°;
3b 95% yield, m.p.130°C, [[alpha]]D -1.1°).
Preparation of [alpha]-Amino Phosphonates (5): A mixture of urea
(1mmol), diethyl phosphite (1.5mmol), and acetyl chloride (5ml) at 0°C
under dry argon, was treated with the aldehyde (1.5mmol) over 10 min. The
reaction mixture was stirred at this temperature for 30 min, and then for 1 hr
at room temperature. Standard extractive workup afforded the products. (refer
to Scheme 1).
Imine Coupling: To a stirred solution of the appropriate
imidazolidin-2-one (1 equiv.) in dry CH2Cl2 (4 ml) at
0°C were added iPr2NEt (1.2 equiv.) and
TiCl4 (1.1 equiv.). The resultant purple enolate was stirred at
0°C for 20 minutes, cooled to 55°C, and treated with a premixture of
TiCl4 (1.5 mmol) and N-(benzylidene) toluene-4-sulphonamide (1.5
mmol) in CH2Cl2 (4 ml). The mixture was stirred at -
55°C for 3h, quenched with saturated aqueous ammonium chloride (20 ml),
warmed to room temperature, and extracted with CH2Cl2
(3x20 ml). The organic phase was dried (MgSO4) concentrated and
purified. (7 R = Me, m.p.210°C, [[alpha]]D -7.8°;
8 R = Me, m.p.242°C, [[alpha]]D -178°; 8 R =
Ph, m.p.262°C, [[alpha]]D -118°)