| [Related articles/posters: 121 053 013 ] |
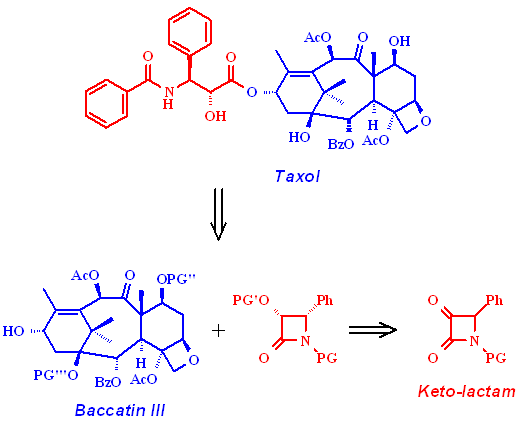
As the production of Taxol is still far from being optimized, there is still a lot of research going on in this field (1). Up to now a large scale total synthesis is lacking and the access by isolation of the bark of the Pacific Yew tree is very limited. The precursor Baccatin III can be isolated from the needles of the Atlantic Yew tree as a quickly regrowable resource. An industrial process takes advantage of the b-lactam method to introduce the side chain into this polycyclic core (2).
Our approach to synthesize this key intermediate is based on the enantioselective
reduction of an a-keto-b-lactam
precursor. We studied several natural, mutant, and engineered yeast strains
for their selectivity in the chiral resolution step involved.
Bakerâs yeast is well established as a bioorganic reducing agent for ketones and ketoesters (3). Its cheap and easy accessibility and the ease of cultivating this microorganism made it popular among organic chemists. Moreover, the use of yeast represents an environmentally compatible technique ö a feature becoming increasingly important ö and the organism is pathogenically benign. Apart from technical problems such as solubility problems of the starting material in aqueous cultures and/or possible toxicity of the substrate to yeast, one of the major downsides of using yeast for reductions is the fact that these reactions are frequently but not always highly enantioselective. In substrates with a preexisting chiral center, enantioselectivity as well as diastereoselectivity may be low. This can be attributed to the presence of several reductases with overlapping substrate acceptabilities (4). In an effort to find a selective reducing agent for a-keto-b-lactams we have studied some natural, mutated, and bioengineered yeast strains.
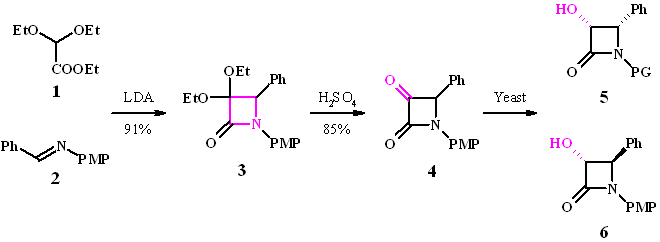
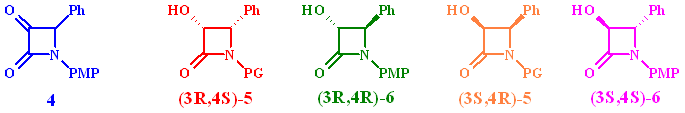
Cyclization of ester 1 with imine 2 in a modified Staudinger reaction using LDA gives the azetidine ring system in excellent yield. Deprotection of the ketal 3 requires rather forcing conditions (75% H2SO4), however yields of the ketone 4 are high.
Four isomers are possible after reaction: the enantiomeric cis products 5 and the trans compounds 6. The required stereoconfiguration for the Taxol side chain is (3R,4S)-5.
Assuming that the si/re-face attack model is valid, the predominantly active enzyme for a-keto-b-lactams seems to be a D-enzyme giving predominantly the R-enantiomer.
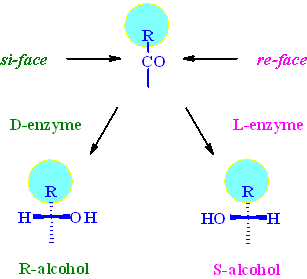
The R-alcohol with the correct stereo configuration for Taxol
is the predominantly formed cis-isomer (3R,4S)-5 in
all reactions. This indicates that the major enzyme system responsible
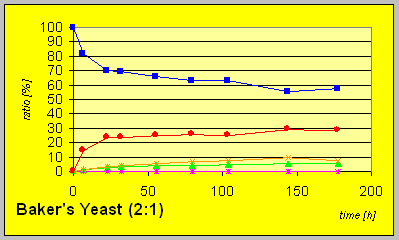
for the biotransformation belongs to the D-family. Plots of the
reaction progress indicate that all strains exhibit resolution of the racemic
starting material 4. Formation of the diastereomeric trans-compound
(3R,4R)-6 is observed after consumption of substantial amounts
of starting material. The yields of the reductions on complete conversion
were quantitative in most cases.
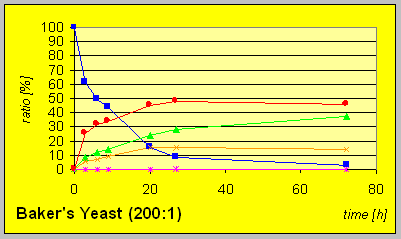
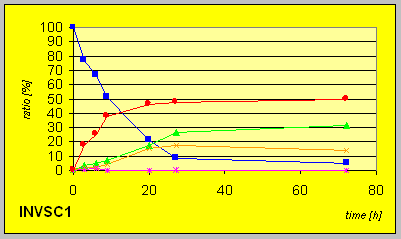
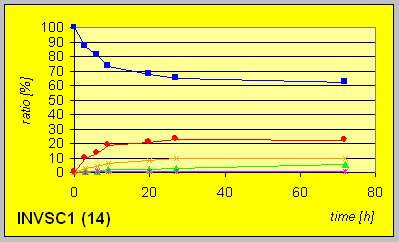
In the laboratory strain INVSC1 the D-reductase seems to be expressed in excess compared to ordinary BakerÎs yeast. Hence much lower amounts of biocatalyst were necessary to give complete conversion in comparable times combined with better resultion capability.
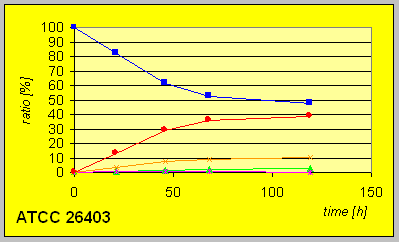
Much slower conversion rates and yields of alcohols were detected with the mutant strain ATCC 26403 with a deficiency in the fatty acid synthetase expression system that carries D-reaductase activity. However, the fact that still reduction to an R-alcohol could be observed indicate the potential presence of another D-enzyme.
The genetically engineered strain INVSC1(pSRG14) overexpressing the aldo-keto reductase L-1 exhibits a slower conversion that can be attributed to the still present activity of the D-enzyme.
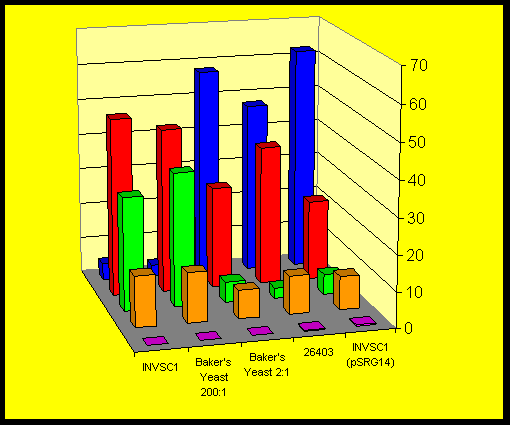
The fact that all yeast strains studied produced similar amounts of the cis-isomer (3S,4R)-5 after comparable times indicate that a-keto-b- lactams might be accepted by another reductase giving S-alcohols. Only minor traces of the corresponding diastereomeric compound (3S,4S)-5 were detected.
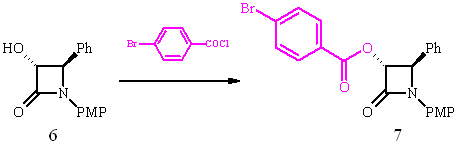
Structural assignment of (3R,4R)-6 was carried out by X-ray diffraction of derivative 7 utilizing the heavy atom effect of bromine to determine the absolute configuration.
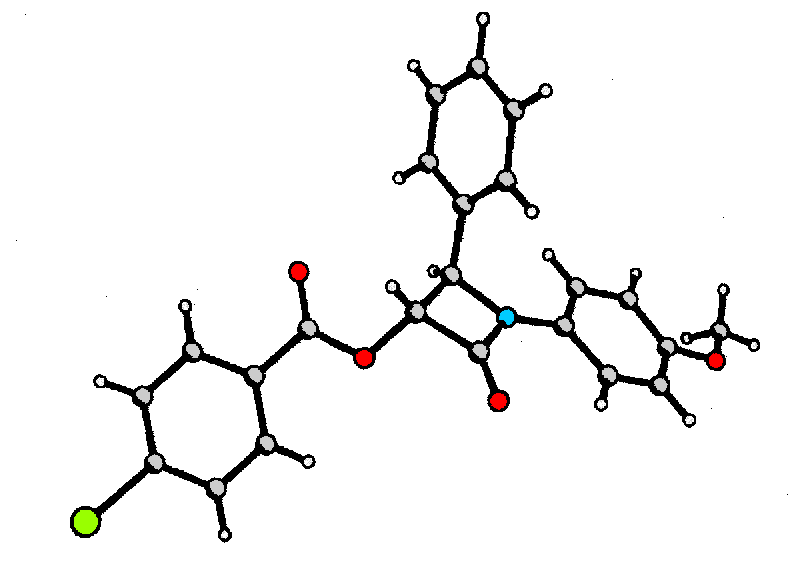

![]() We would
like to thank Dr. Fernande D. Ronchon, Département de chimie, Université
du Québec à Montréal, for performing X-ray diffraction
and structure elucidation of compound 7.
We would
like to thank Dr. Fernande D. Ronchon, Département de chimie, Université
du Québec à Montréal, for performing X-ray diffraction
and structure elucidation of compound 7.