The reported methodology has been further investigated in order to provide a new efficient entry towards unsaturated 2â,3â-dideoxynucleosides as d4T and its analogues. The interest for this synthetic way is widely demonstrated by the known and clinically tested anti-HIV activity of d4T. Thus d4T 55 and its methyl analog 57 have been synthesized after removal of the N-dimethylamino group in 48a and 53 respectively, performed, according to a Cope elimination, by treatment with MCPBA (60-65% yield), followed by TBAF treatment (Scheme 10).
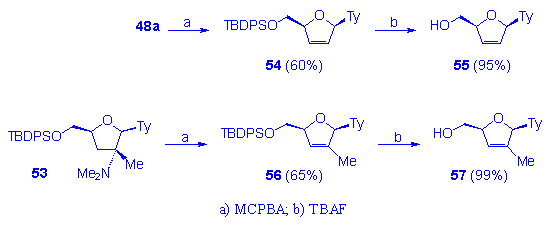
Scheme 10
The alternative possibility to synthesize 55 and 57, by introduction of the unsaturated bond before the coupling reaction with the silylated thymine, has been explored with unsuccessful results. In fact, DIBALH reduction of the butenolide 59, obtained via Cope elimination from 58, afforded the compound 60 which on further acetylation furnished the tert-butyl[(4-methyl-2-furyl)methoxy]diphenylsilane 61 (Scheme 11).
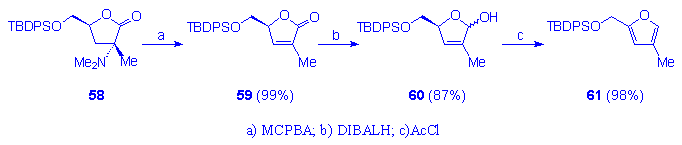
Scheme 11
The result can be explained according to the consideration that 2,3-unsaturated pentafuranosides are sensitive to both acidic and basic conditions, easily affording furan derivatives. The reported procedure for the synthesis of d4T is, however, weakened by the consideration that the acetyl precursor 47a (Figure 8) is obtained only as a minor product during the DIBALH stereoselective reduction of the cis lacton 45 (46a/46b ratio = 1:3). According to these considerations, we have overcome the problem by performing the epimerization of 45 with DBU, followed by DIBALH reduction which afforded a mixture of 63a and 63b (3:1 ratio). After conversion into the corresponding acetyl derivatives 64a and 64b, the acetyl derivative 64a has been separated and then transformed in d4T through the 2â-dimethylamino nucleoside 65a, via Cope elimination. Moreover, 65a can be also converted in 66a by reaction with TBAF (Scheme 12).
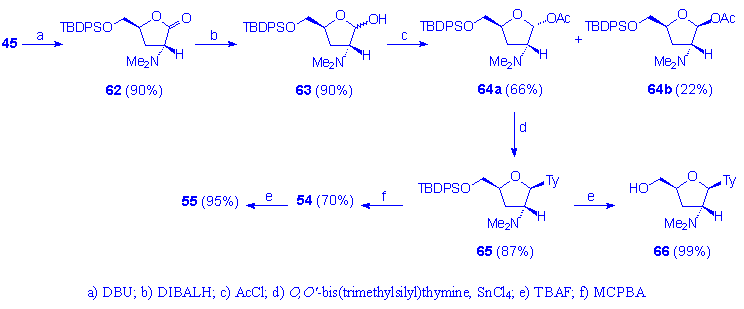
Scheme 12
In conclusion, the novel [3+2] cycloaddition methodology outlined herein provides a general and efficient access to modified N,O-nucleosides, 2â-dimethylamino and 2â,3â-dideoxynucleosides. The designed scheme constitutes an excellent alternative to previously reported approaches and shows its versatility in the potential construction of other nucleosides with different purine and pyrimidine bases. Furthermore, hydrogenation of the 2â,3â-double bond could represent an alternative synthetic pathway to saturated dideoxynucleosides as DDC, DDI, 3TC. Exploitations of the scope and potential of this synthetic scheme, in the aim of the obtainment of optically active dideoxynucleosides, are also in progress.
Acknowledgment
The autors are grateful to Prof. Giovanni Purrello for stimulating discussion and to M.U.R.S.T. for its financial support.
![]() Synthesis
of 2â-dimethylamino-2â,3â-dideoxynucleosides
Synthesis
of 2â-dimethylamino-2â,3â-dideoxynucleosides
![]() Synthesis of d4T and
its methyl analogue
Synthesis of d4T and
its methyl analogue