| [Related articles/posters: 112 013 121 ] |

However, we were attracted to this natural 2-chloropyridine, because it offers high potential for chemical modification by substitution of the non-pharmocophoric chloride. Particularly the published, facile elaboration of 2-halopyridines to 2,2‰-bipyridines [29-38] should give novel chelators with an additional chiral amino function in the side chain.
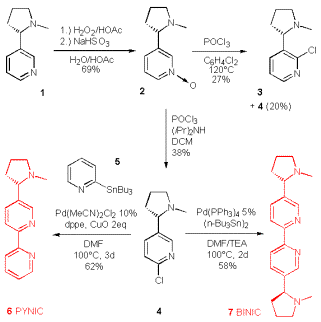
We initially sketched a synthesis of a pyridyl-substituted epibatidine, which may coordinate to metals with different ligation preferences in a helical manner as depicted in Scheme 2. However for ease of synthesis we switched to the simpler 6-chloronicotine as a precursor for our bipyridyls, because it is accessible in 2 steps from nicotine (Scheme 3).
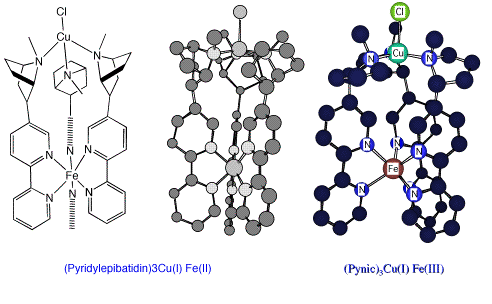
Pyridyl epibatidine Fe(II)/Cu(I) and PYNIC Fe(III)/Cu(I) complexes modelled in MM2 (ChemDraw 3D)
Thus (-)-nicotine was converted into the pyridine N-oxide by Taylor‰s method [39] and then treated with POCl3 to give the isomeric chloronicotines 3 and 4. Addition of diisopropylamine overrides the steering effects of the neighbouring nitrogen and results in the exclusive formation of the desired isomer 4, albeit at low overall yield [40]. This regiocontrol of a 2Á-amine is unique for diisopropylamine, all other tested 2Á amines lead to isomeric mixtures.
We assume the competition of the amines for the highly reactive POCl3 results in the formation of an ammonium phosphoryl species, which in the case for nicotine results in the short path chloride transfer via the Boekelheide reaction [36, 41]. The sterically more demanding diisopropylamine gives rise to a selective reagent, which avoids the steric strain involved in the substitution at the 2-position.
The 6-chloronicotine was then converted into either BINIC (6) or PYNIC (7) by a palladium mediated cross-coupling using either 2-tri-n-butylstannylpridine or hexa-butylditin. Both reaction proceeded smoothly in DMF, although in just 58 and 62 yield. Whereas additional copper oxide improved the yield for PYNIC from 52 to 62%, the yield of BINIC was not improved by any further additives or ligands: Cu(I), Cu(II), TBACl, dppe, dppp.
Unfortunately the obtained 2,2‰-bipyridyls stabilise copper(II) and do not give stable Cu(I) helical complexes. They result in the rapid disproportionation of Cu(I) to Cu and Cu(II) and thus form amorphous Cu(II) precipitates. On the other hand the simpler and stable palladium(II) and mercury(II) complexes 8 and 9 are obtained by precipitation from acetonitril and recrystallization.
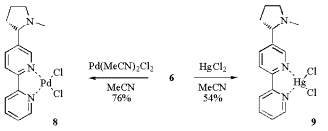
Scheme 4
We therefore turned our attention to related chiral bipyridyls with reduced donor capacity in the side chains. Amides and esters are less likely to cause the disproportionation of Cu(I) and are easily accessed by condensation of 6-chloronicotinoylchloride (10) and amino acids such as proline methylester.
This nicotine amide was dimerized to the bipyridine under non-optimised conditions at just 12% yield. The improvement of this reaction by replacement of hexabutylditin by the superior reagent hexamethylditin [42] is subject of ongoing investigations.
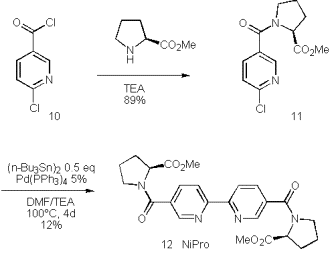
Scheme 5
Another potential for chiral bipyridyl complexes may be found in the preparation of defined 2- and 3-dimensional assemblies such as micelles, nanotubes and monolayered films.
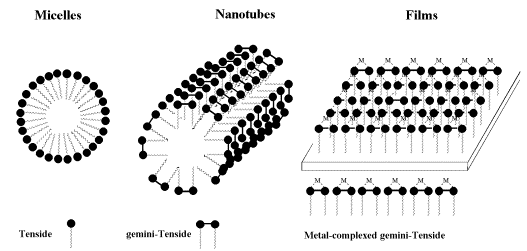
Scheme 6
Gemini-surfactants [43, 44] are predicted to form nanotubes, the incorporation of bipyridyls into amphiphiles may yield such desired nanotubes. To this end we have started the synthesis of the benzyl protected dodecyl-a(D)-glucosides, which starts form anomerically pure dodecyl a(D)-glucoside (13), which is protected at the 6-position by tritylchloride in pyridine.
Benzylation of the 2,3,4-positions by benzylchloride and detritylation by heating the tetraether in ethanol in the presence of pyridine*hydrochloride renders the glucose derivative 14 ready for a selective 6-esterification by 2,2‰-bipyridine-4,4‰-dicarboxylic acid or 6-chloronicotinic acid via the corresponding acid chlorides 15 and 16.
The short route via the rather expensive bipyridine 16 gives the product in 70% yield, but this approach is hampered by the low solubility and the unreliable synthesis of the acid chloride 16. However, the esterification of the alcohol 14 by inexpensive 6-chloronicotinoylchloride proceeds quantitatively and gives the product upon treatment with Pd(PPh3)4 and hexabutylditin in similar yield. Again the deprotection, complexation and physical properties of this ligand are subject of ongoing investigations.
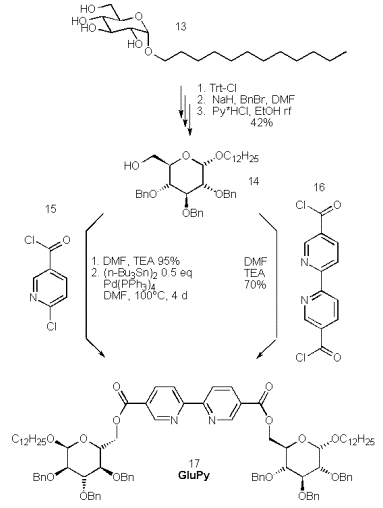
Scheme 7
The complexation of transition metals by amino acids is a well documented way [45, 46] to control shape and activity of peptides and proteins. On our way [47] to synthetic kinase inhibitors, kinase markers and designed haloperoxidases [48, 49] we are interested in 4-substituted pyridine boxes with two donor systems adjacent to the pyridyl nitrogen.
Alteration of the donor and pH allows selective coordination to a range of cations such as Cu(I), Zn(II), Ln(III), V(V) and Tc(VII). The incorporation of these complexes into peptides may give access to peptide mimetics or enzyme inhibitors with new or improved activity. The alterations in polarity, geometry and additional vacant ligation sites hold potential to control peptide secondary and tertiary structure.
Other applications may be found in identification and purification of synthetic peptides by Immobilised Metal Affinity Chromatography (IMAC) [50].
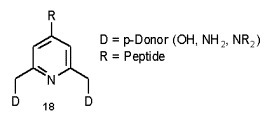
Scheme 8
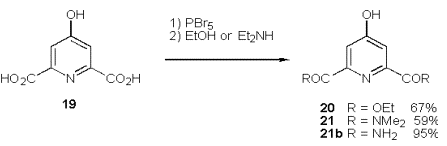
On our way to such peptide linked ligands (18, Scheme 1) we follow Takalo‰s route [51] to 2,6 substituted 4-bromopyridines. The procedure provides a variety of diesters and diamides by reactions of the intermediate dibromide with alcohols or amides (Scheme 9).
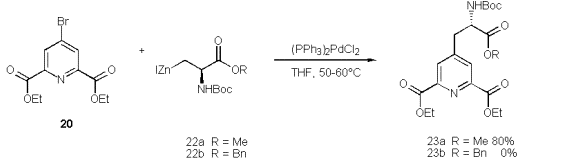
As amino building block we use JacksonÇs versatile zinc reagents [52] 22a, 22b which are synthesised from Boc protected serine esters by tosylation, substitution with NaI, followed by treatment with activated zinc. This zinc mediated cross-coupling reaction of iodoserines has been proven to be the method of choice.
Scheme 10
The zinc is activated by KnochelÇs method [53] (BrCH2CH2Br, TMSCl/THF) to form 22a within 1.5h to 2h. It can be separated easily from excess zinc via syringe and transferred to a 2nd round bottom flask containing 20 and (Ph3P)2PdCl2. A dark brown solution results, which is heated to 35ÁC for 24h [47].
In accord to Walker‰s observation [54] we found that the zinc reagent of iodoserine methylester forms more rapidly than that of the corresponding benzylester. The extended reaction time required to transform the benzyl derivative favours an elimination to give didehydroalanines from residual starting material. Walker assigned the difference in the reaction rates of 23a and 23b to chelation control.
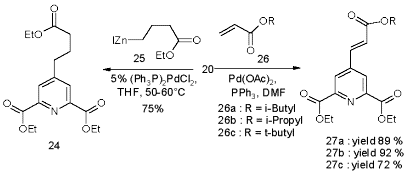
The more desirable 23b, because of it‰s selective deprotection, is not accessible by this coupling method. Yet the methylester 22a couples readily and at good yields, but a selective deprotection of the methyl group in 23a met with failure. A simplified analogue derives from zinc reagent 25 to give the propionate 24 in satisfactory yield. However, the alternative Heck olefination [55] of acrylates 26a-c by 20 turns out to be far easier, and provides higher yields of the acrylates 27a-c.
Scheme 11
The corresponding acid is limited to N-terminal capping of peptides, but offers a convenient synthesis by Heck olefination of the bromopyridine 20 with inexpensive acrylates to give the triesters 27a-c in good to excellent yields under simple, non inert reaction conditions.
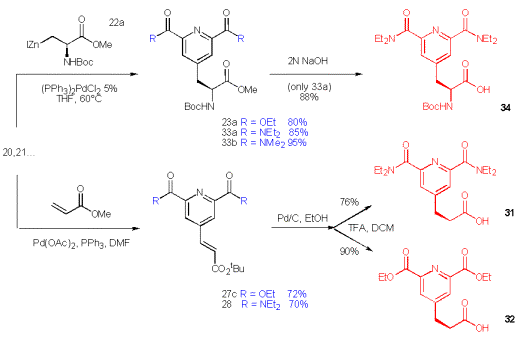
The synthesis of a new chelating pyridine 34, which is suited for Solid Phase Peptide Synthesis (SPPS) is viable for the 2,6-diamide 21. Following the same approach the methylester 33 is obtained and subsequently hydrolysed to the free acid 34, which is ready for SPPS or batch synthesis of peptides.
Scheme 12
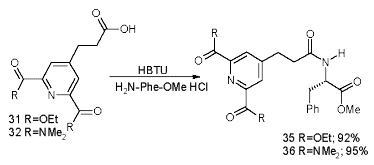
We established the complexation site of our carboxylated and bisamidated chiral and non-chiral ligands coupled to phenylalanine by titration with 0.1 to 3.2 eq of Eu(III), monitored by 1H-NMR. Whereas the signals for the phenylalanine part remain unchanged, the signals for the aromatic proton and donor groups shift towards a maximum (Scheme 14).
Scheme 13
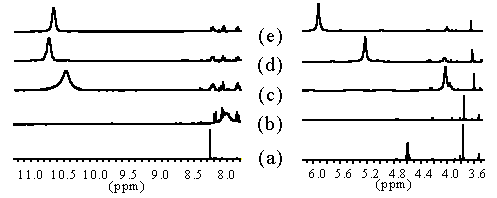
Europium-shift measurements. All samples in MeCN-D3, 400 MHz (a) 35, (b) + 0.12 eq Eu(NO3)3, (c) + 1.2 eq Eu(NO3)3, (d) + 1.6 eq Eu(NO3)3, (e) + 2.3 eq Eu(NO3)3.
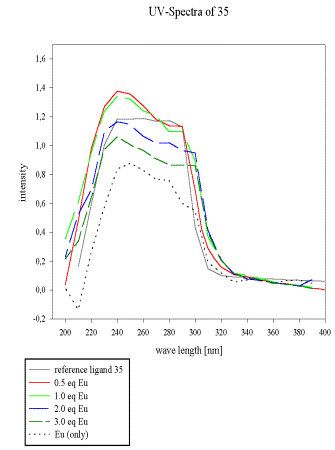
All samples in MeCN, reference (0.01M), 0.5 eq, 1 eq, 2 eq, 3 eq of Eu(NO3)3 x 6 H2O.
The UV-spectra of the acrylate 28 and the pseudopeptide 35 with a similar 2,6-bis(diethylamido)pyridine subunit and their dependence on europium(III) concentration was compared to evaluate the contribution of the unsaturated acrylate to the total absorbency spectrum of 28.
This is of particular interest for non-radioactive labelling of bioactive material by europium fluorescence. A weak complexation of 35 and Eu(III) is observed in acetonitrile, which results in enhanced extinction between 220 and 260 nm.
As soon as more than 1 eq of europium/ligand is added, a new species becomes predominant and significantly alters the UV-absorbency (Scheme 15). The acrylate 28, by comparison, is a better ligand for Eu(III), which is reflected by its UV-spectrum as depicted in Scheme 16.
The continuous decrease of the ligand spectrum upon Eu(III) addition points to strong initial complexation. Remarkably, no new bands are observed for the formed complex within the indicated wavelength range.
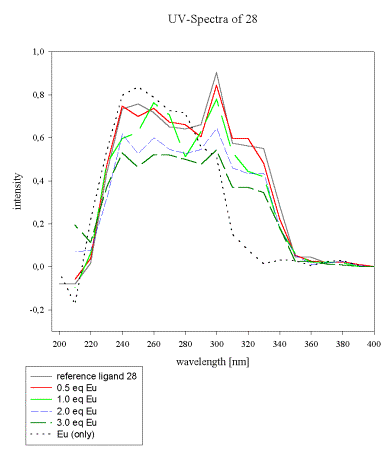
All samples in MeCN, reference (0.01M), 0.5eq, 1eq, 2eq, 3eq of Eu(NO3)3 x 6 H2O
Summary
We have synthesised a number of pyridines and 2,2'-bipyridines with additional chiral side-chains to induce chirality in metal-chelates and higher assemblies thereof. The helical nature of europium complexes of the ligands 30 and 33 is subject of a future communication in collaboration with C. Piguet. The helical Ru(II) complexes of the 2,2'-bipyridines are under current investigation.
[2] S. R. Fletcher, R. Baker, M. S. Chambers, S. C. Hobbs, P. J. Mitchell, J. Chem. Soc., Chem. Commun. 1993, 1216.
[3] K. Senokuchi, H. Nakai, M. Kawamura, N. Katsube, S. Nonaku, H. Sawaragi, N. Hamanaka, Synlett 1994, 343.
[4] E. J. Corey, T.-P. Loh, S. AchyuthaRao, D. C. Daley, S. Sarshar, J. Org. Chem. 1993, 58, 5600.
[5] C. A. Broka, Tetrahedron Lett. 1993, 34, 3251.
[6] S. C. Clayton, A. C. Regan, Tetrahedron Lett. 1993, 34, 7493.
[7] D. F. Huang, T. Y. Shen, Tetrahedron Lett. 1993, 34, 4477.
[8] K. Sestanj, E. Melenski, I. Jirkovsky, Tetrahedron Lett. 1994, 35, 5417.
[9] S. Y. Koo, J. Lerpiniere, I. D. Linney, R. Wrigglesworth, J. Chem. Soc., Chem. Commun. 1994, 1775.
[10] E. Albertini, A. Barco, S. Benetti, C. De Risi, G. P. Pollini, R. Romagnoli, V. Zanirato, Tetrahedron Lett. 1994, 35, 9297.
[11] P. L. Kotian, F. I. Caroll, Synth. Commun. 1995, 25, 63.
[12] G. Pandey, T. D. Bagul, G. Lakshmaiah, Tetrahedron Lett. 1994, 35, 7439.
[13] K. Okabe, M. Natsume, Chem. Pharm. Bull. 1994, 42, 1432.
[14] E. V. Dehmlow, J. Prakt. Chem. 1995, 337, 167.
[15] R. Xu, G. Chu, D. Bai, Tetrahedron Lett. 1996, 37, 1463.
[16] C. Szántay, Z. Kardos-Balogh, I. Moldvai, C. Szántay Jr., E. Temesvári-Major, G. Blaskó, Tetrahedon 1996, 52, 11053.
[17] B. M. Trost, G. R. Cook, Tetrahedron Lett. 1996, 37, 7485.
[18] G. M. P. Giblin, C. D. Jones, N. S. Simpkins, Synlett 1997, 589.
[19] D. Bai, R. Xu, X. Zhu, Drugs of the Future 1997, 22, 1210.
[20] Z. Chen, M. L. Trudell, Tetrahedron Lett. 1994, 35, 9649.
[21] K. Hiroya, K. Uwai, K. Osagawara, Chem. Pharm. Bull. 1995, 43, 901.
[22] J. R. Malpass, D. A. Hemmings, A. L. Wallis, Tetrahedron Lett. 1996, 37, 3911.
[23] A. L. E. Larsson, R. G. P. Gatti, J.-E. Bäckvall, J. Chem. Soc., Perkin Trans. I 1997, 2873.
[24] N. P. Pavri, M. L. Trudell, Tetrahedron Lett. 1997, 38, 7993.
[25] Z. Chen, M. L. Trudell, Chem. Rev. 1996, 96, 1179.
[26] R. Xu, D. Bai, G. Chu, J. Tao, X. Zhu, Bioorg. Med. Chem. Lett 1996, 6, 279.
[27] D. Barlocco, G. Cignarella, D. Tondi, P. Vianello, S. Villa, A. Bartolini, C. Ghelardini, N. Galeotti, D. J. Anderson, T. A. Kuntzweiler, D. Colombo, L. Thoma, J. Med. Chem. 1998, 41, 674.
[28] A. W. Bannon, M. W. Decker, M. W. Hollady, P. Curon, D. Donelly-Roberts, P. S. Puttfarcken, R. S. Bitner, A. Diaz, A. H. Dickenson, R. D. Porsolt, M. Williams, S. P. Arneric, Science 1998, 279, 77.
[29] J. A. Zoltewicz, M. P. Cruskie Jr., Tetrahedon 1995, 51, 3103.
[30] V. N. Kalinin, Synthesis 1992, 413.
[31] M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli, M. Montanucci, Synthesis 1984, 736.
[32] R. Vanderesse, M. Lourak, Y. Fort, P. Caubere, Tetrahedron Lett. 1986, 27, 5483.
[33] P. Bamfield, P. M. Quan, Synthesis 1978, 537.
[34] G. S. Hanan, J.-M. Lehn, N. Kyritsakas, J. Fischer, J. Chem. Soc., Chem. Commun. 1995, 765.
[35] N. Murata, T. Sugihara, T. Sakamoto, Synlett 1997, 298.
[36] J. A. Zoltewicz, M. P. Cruskie Jr., C. D. Dill, Tetrahedon 1996, 52, 4239.
[37] J. A. Zoltewicz, M. P. Cruskie Jr., Tetrahedon 1995, 51, 11393.
[38] J. A. Zoltewicz, M. P. Cruskie Jr., Tetrahedon 1995, 51, 11401.
[39] E. C. Taylor, N. E. Boyer, J. Org. Chem. 1959, 24, 275.
[40] B. Schmidt, V. Neitemeier, Synthesis 1998, 42.
[41] C. Fontenas, E. Bejan, H. Ait Haddou, G. G. A. Balavoine, Synth. Commun. 1995, 25, 629.
[42] M. Benaglia, S. Toyota, C. R. Woods, J. S. Siegel, Tetrahedron Lett. 1997, 38, 4737.
[43] M. J. L. Castro, J. Kovensky, A. F. Cirelli, Tetrahedron Lett. 1997, 38, 3995.
[44] S. Karaborni, K. Kesselink, P. A. J. Hilbers, B. Smit, J. Karthäuser, N. M. van Os, R. Zana, Nature 1994, 266, 254.
[45] B. Imperiali, S. L. Fisher, J. Am. Chem. Soc. 1991, 113, 8527.
[46] C. R. Palmers, L. S. Sloan, J. C. Adrian Jr., B. Cuenoud, D. N. Paolella, A. Schepartz, J. Am. Chem. Soc. 1995, 117, 8899.
[47] B. Schmidt, D. K. Ehlert, Tetrahedron Lett. 1998, 39, 3999.
[48] S. L. Neidleman, J. Geigert, Biohalogenation: Principles, Basic Roles and Applications, Ellis Horwood Limited, Chichester, 1986.
[49] G. J. Colpas, B. J. Hamstra, J. W. Kampf, V. L. Pecoraro, J. Am. Chem. Soc. 1996, 118, 3469.
[50] G. N. Lindeberg, H. Bennich, Å. Engström, Int. J. Pept. Protein Res. 1991, 38, 253.
[51] H. Takalo, P. Pasanen, J. Kankare, Acta Chem. Scand. Ser. B 1988, 42, 373.
[52] R. F. W. Jackson, J. Org. Chem. 1992, 57, 3397.
[53] S. AchyuthaRao, P. Knochel, J. Org. Chem. 1991, 56, 4591.
[54] M. A. Walker, K. P. Kaplita, T. Chen, H. D. King, Synlett 1997, 169.
[55] A. Spencer, J. Organomet. Chem. 1983, 258, 101.