| [Related articles/posters: 015 097 117 ] |
Introduction
Dihydropyrimidines of type 1 (DHPMs, Biginelli compounds) [1] represent a long-known class of heterocycles that show a diverse range of biological activities [2]. Most notable is the recently discovered cardiovascular (i.e. antihypertensive) activity exhibited by certain functionalized derivatives, e.g. 2, that compares very favorable with the activity displayed by the structurally related antihypertensive dihydropyridine drugs (DHPs) amlodipine and nicardipine (e.g. 3) [3,4]. Although several questions about the exact stereochemical and conformational requirements for activity remain, a reasonable pharmacophore model for DHP/DHPM calcium channel modulators has been proposed recently [5]. The main features of this model include: (i) a boat-like conformation of the DHP/DHPM ring; (ii) an axially positioned substituted aryl ring and (iii) an ester group at C5 oriented cis with respect to the C5=C6 double bond [5].
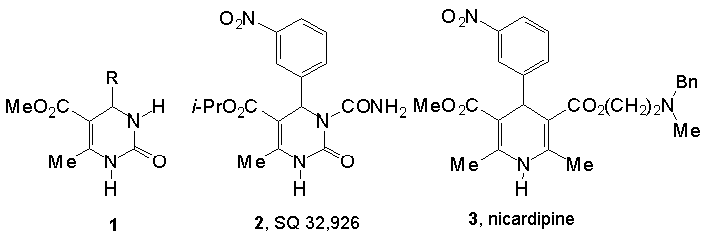
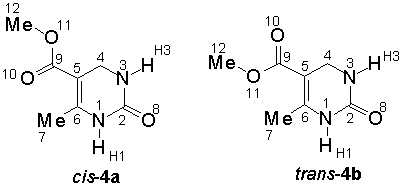
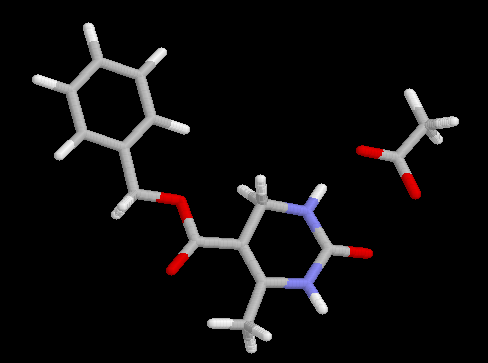
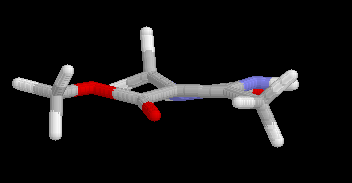
Relative energies and the torsional angle j (C6-C5-C9-O10), characterizing the ester conformation (cis ca. 0°, trans ca. 180°) for the two main rotamers of 4a,b are summarized in Table 1. Independent of the basis set as well as the computational level (RHF vs. B3LYP vs. MP2) invariably a slight preference for the cis rotamer 4a is predicted by the calculations, indicating an intrinsic greater stability of this isomer. The energy difference, however, is quite small (ca. 1 kcal/mol) and is decreasing with an increasing level of theory (B3LYP yield nearly identical E values as RHF, those obtained by MP2 are slightly smaller). It is, therefore, not surprising that one can observe experimentally a rather subtle dependence of the preferred conformation of such DHPMs (i.e. 1, R = Ar) on structural modifications of the parent molecule [6]. The benzyl ester derivative 5 adopts the cis conformation in the solid state (Figure 1). Intermolecular interactions, e.g. hydrogen bonding of the ester carbonyl CO to the N1-H of a second DHPM molecule in the solid state [6], may preferentially stabilize the trans rotamer, overriding the inherent preference for the cis ester orientation. In both conformations, the ester group is nearly coplanar to the C5=C6 double bond with the largest twist angles (< 12°) obtained at the MP2 level and the smallest one (< 5°) with B3LYP. In the solid state, for benzyl ester 5, the corresponding value is -2.3°. For the 4-aryl derivatives 1 (R = Ar) ester torsions of 1.3° and 12.6° for cis, and -165.4° and -166.4° for trans isomers, respectively, are found by X-ray structure determination [6]. Calculated bond lenghths for DHPMs 4a,b have been compared with the distances obtained by X-ray crystallographic analysis of 5. There is a reasonable good agreement between the calculated and experimental values.

In the present poster we have presented
a detailed ring conformational analysis of dihydropyrimidine 4 based
on ab initio and density functional calculations at various levels
of theory. While all experimental data - at least in the solid state -
indicate an almost completely planar six-membered ring in C4-unsubstituted
DHPMs [7], the computations suggest significant puckering (Figure
2). However, we find a strong dependence of the amount of puckering on
both the basis set (inclusion of polarization functions inevitably leads
to puckered structures) as well as the computational method (increasing
deviation from planarity in the order RHF < B3LYP < MP2).
An analogous sensitivity on the employed computational procedure had been
observed previously in amides and ureas [9-16]. While in simple
amides and ureas such an effect is reflected in differences of amide bond
planarity, in cyclic derivatives like 4, puckering of the six-membered
ring will be observed. Whether the nonplanarity of the dihydropyrimidine
ring in 4 is an intrinsic property of the molecule itself or an
artefact of the calculation cannot be decided with certainty at this point:
the computed energy differences between planar and puckered ring geometries
are rather small (Table 3). Therefore, intermolecular
forces (crystal packing) might lead to at least some flattening. On the
other hand, difficulties of MP2 calculations in the correct treatment of
conformational properties of amino acids [17] and
amides (overestimation of the deviation of the heavy atom skeleton from
planarity) [18] are known. Pending gas phase structure
and puckering potential measurements at present no definitive conclusions
concerning the intrinsic ring conformation of dihydropyrimidines are possible.
Acknowledgements
This work was supported by the Austrian
Academy of Sciences and the Austrian
Science Fund. We thank Dr. Marcus A. Semones for obtaining X-ray data
of compound 5.
[1] P. Biginelli, Gazz. Chim. Ital., 23, 360 (1893).
[2] For a review, see: C. O. Kappe, Tetrahedron, 49, 6937 (1993).
[3] (a) K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, D. Moreland, A. Hedberg, and B. C. O'Reilly, J. Med. Chem., 34¸806 (1991); (b) K. S. Atwal, G. C. Rovnyak, S. D. Kimball, D. M. Floyd, S. Moreland, B. N. Swanson, J. Z. Gougoutas, J. Schwartz, K. M. Smillie, and M. F. Malley, J. Med. Chem., 33, 2629 (1990).
[4] G. C. Rovnyak, K. S. Atwal, A. Hedberg, S. D. Kimball, S. Moreland, J. Z. Gougoutas, B. C. O'Reilly, J. Schwartz, and M. F. Malley, J. Med. Chem., 35, 3254 (1992).
[5] G. C. Rovnyak, S. D. Kimball, B. Beyer, G. Cicinotta, J. D. DiMarco, J. Gougoutas, A. Hedberg, M. Malley, J. P. McCarthy, R. Zhang, and S. Moreland, J. Med. Chem., 38, 119 (1995). For a discussion, see: D. J. Triggle, and S. Padmanabhan, Chemtracts: Org. Chem., 8, 191 (1995). For previous models, see: S. Goldmann and J. Stoltefuss, Angew. Chem., Int. Ed. Engl., 30, 1559 (1991).
[6] C. O. Kappe, W. M. F. Fabian, and M. A. Semones, Tetrahedron, 53, 2803 (1997).
[7] Apart from the X-ray analysis of 5 reported herein, a CSD search revealed only two other X-ray structures of C4-unsubstituted DHPM derivatives, also yielding almost planar ring geometries: (a) SOBDIW: Ethyl-4,5,6,7-tetrahydro-4-methyl-5-oxo-2H-pyrazolo(4,3-d)pyrimidine-2-carboxylate: C. O. Kappe and G. Färber, J. Chem. Soc., Perkin Trans. 1, 1342 (1991); (b) VICGUJ: 1-Phenyl-5-cyanomethyl-4,5,6,7-tetrahydro-1H-pyrazolo(3,4-d)pyrimidin-6-one: R. Ferroni, D. Simoni, P. Orlandini, M. Guarneri, V. Bertolasi, and V. Ferretti, J. Heterocycl. Chem., 27, 823 (1990).
[8] W. J. Adams, H. J. Geise, and L. S. Bartell, J. Am. Chem. Soc., 92, 5013 (1970).
[9] (a) H. M. Sulzbach, G. Vacek, P. R. Schreiner, J. M. Galbraith, P. von R. Schleyer, and H. F. Schaefer III, J. Comput. Chem., 18, 126 (1997). (b) H. M. Sulzbach, P. von R. Schleyer, and H. F. Schaefer III, J. Am. Chem. Soc., 117, 2632 (1995).
[10] H.-G. Mack, H. Oberhammer, J. Am. Chem. Soc., 119, 3567 (1997).
[11] G. M. Wright, R. J. Simmonds, and D. E. Parry, J. Comput. Chem., 9, 600 (1988).
[12] M. Kontoyianni and J. P. Bowen, J. Comput. Chem., 13, 657 (1992).
[13] M. W. Wong and K. B. Wiberg, J. Phys. Chem., 96, 668 (1992).
[14] J. E. Boggs and N. V. Riggs, J. Comput. Chem., 6, 46 (1985).
[15] B. Kallis and R. Mitzner, J. Chem. Soc., Perkin Trans. 2, 1397 (1996).
[16] G. Rasul, G. K. S. Prakash, and G. Olah, J. Org. Chem., 59, 2552 (1994).
[17] R. F. Frey, J. Coffin, S. Q. Newton, M. Ramek, V. K. W. Cheng, F. A. Momany, and L. Schäfer, J. Am. Chem. Soc., 114, 5369 (1992).
[18] K.-M. Marstokk, H. Møllendal, and S. Samdal, J. Mol. Struct., 376, 11 (1996).