| [Molecules: 46] [Related articles/posters: 024 001 063 048 052 ] |
We wish to present here, a summary of our recent results in the syntheses of
natural and/or biologically active molecules in the aza-carbazole,
benzonaphthyridine and pyrido[k,l]acridine series.
Aza-carbazoles
Numerous natural alkaloids belong to the carboline series3 (mainly
carbolines) and display interesting biological properties (harman
derivatives,4 eudistomins,5 lavendamycin,6
picrasma javanica alkaloids7). Until now most syntheses have used
indole or its derivatives as starting materials and are based on condensation
reactions8 between tryptophan or tryptamine and aldehydes.
Substituted compounds are often prepared from available carboline reagents
such as norharman or harman through specific pathway.9 In 1993, we
published10a a general and highly convergent synthesis of the four
parent carbolines 4 based on metalation and cross-coupling reactions
followed by a cyclization (Scheme 1).

Scheme 1
This strategy was successfuly extended to the preparation of -substituted--carbolines10b-e (Scheme 2).
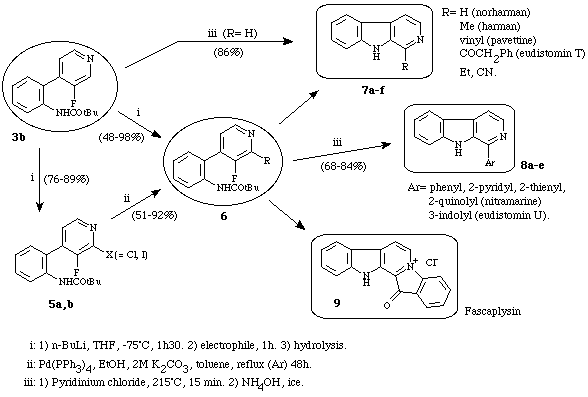
Scheme 2
In such a pathway (partially discussed in a previous paper11), it was found that heterobiaryl 3b was the key molecule of the strategy. Indeed, either by metalation or by metalation/cross-coupling followed by a cyclization, numerous substituted--carbolines could be synthesized from this compound. Most of the prepared carbolines are biologically active molecules (harman, 1-ethyl--carboline, pavettine, nitramarine, fascaplysin and eudistomin T).
The methodology was extended to the preparation of more complex carbolines, i.e. polysubstituted carbolines. First, two models of lavendamycin were made12 (Scheme 3). Thus, the cross-coupling reaction between 1 and 10 regioselectivity gave the 4-phenylpyridine 11. The palladium catalysed cross-coupling reaction between biaryl 11 and 2-quinolylstannane or 5,8-dimethoxy-2-quinolylstannane led to the intermediary triaryl compound 12 (a or b) which was readily cyclized into -carbolines 13 (a or b). Ultimately, the oxidation of 13b (R= OH) with the Fremy's salt afforded the quinoline-5,8-dione 13c in very good yield.
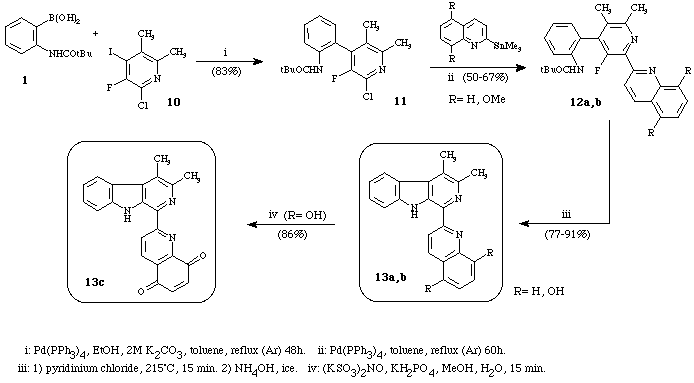
Scheme 3
Then, we prepared13 various hydroxy--carbolines using anisidines as convenient tools for such derivatives (Scheme 4). Arylboronic acids 14a-d were prepared by metalation-boronation of the corresponding anisidines protected as a pivaloyl. The palladium catalysed cross-coupling reaction between phenylboronic acids 14a-d and 3-fluoro-4-iodopyridine 2a using the Suzuki conditions gave the corresponding heterobiaryls 15a-d in good yields. The phenylpyridines were cyclized by treatment with boiling pyridinium chloride to give hydroxy carbolines 16a-d.
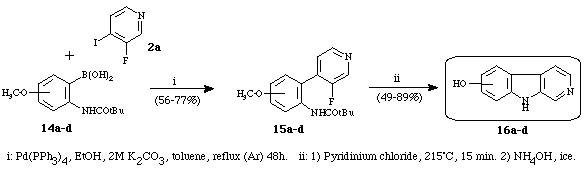
Scheme 4
The strategy has been succesfully extended to the total synthesis13 of the major cytotoxic agent of "cribricellina cribaria": 1-vinyl-8-hydroxy--carboline (Scheme 5).
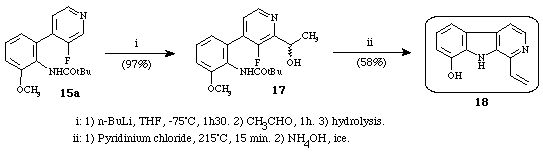
Scheme 5
It was also shown that 6-hydroxy--carboline 16b was subjected to bromination by triphenylphosphine dibromide to give 5-bromo-6-hydroxy--carboline 1910e (eudistomin D) (Scheme 6).
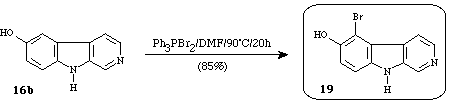
Scheme 6
We also reported the first total synthesis14b of Bauerine B and its demethyl analogue (Scheme 7). Bauerine B (active against HSV-2) is one of three substances isolated in 1994 by Moore et al.14a from the terrestrial blue-green alga Dichothrix baueriana GO-25-2. During the palladium catalysed cross-coupling between boronic acid 20 and iodopyridine 2a, the pivaloyl group was partly hydrolysed. The observed hydrolysis can be explained by the electron withdrawing effect of the two chlorine atoms on the benzene ring which decreases the stability of the amide bond.

Scheme 7
The syntheses of carbolines 16a-d, 18 and 23 illustrate the power of this strategy: any aromatic group (pyridine or benzene) can bear one, two or more substituents before cross-coupling and almost any polysubstituted carboline can be prepared via such a route.
This strategy used in association with the halogen-dance reaction15 allowed us to synthesize carboline and 4-methyl--carboline in a highly convergent route (Scheme 8).
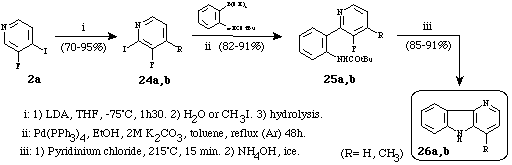
Scheme 8
Ascididemine (27), amphimedine (28), cystoditins (29) and meridine (30) are representative of these series (Scheme 9).
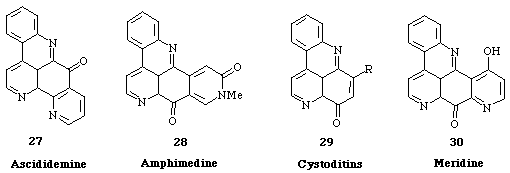
Scheme 9
These natural tetracyclic molecules contain a common structural feature, a 4,5-disubstituted benzo[c]-2,7-naphthyridine ring system which could be useful for the synthesis of the natural tetra- or penta-cyclic molecules (Scheme 10). In the context of combined directed ortho metalation-cross coupling strategies, we developed a new route to benzonaphthyridines and subsequently to natural products or analogues.15, 17
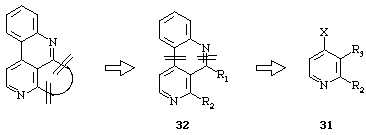
Scheme 10
In accordance with the retrosynthetic pathway depicted above suitable 2,3,4-trisubstituted pyridines 31 are required. Pyridines 31 have been synthesized following two different pathways :
The first way involves ortho-directed metalation of 3-N,N-diisopropylcarboxamido-2-methoxypyridine (33). 9-Methoxy-BBN as well as iodine were used as electrophiles affording pyridines 31a and 31b respectively (Scheme 11).
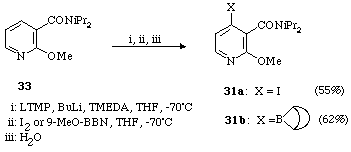
Scheme 11
The second way consists of the metalation of 2-substituted-3-iodopyridine (34a-c).15,17b,c In 1993, we established15 that metalation of iodopyridines by LDA at low temperature was feasible although the pyridine nucleus bears a second halo (fluoro or chloro) substituent. In most cases, lithiation of haloiodopyridines was regioselectively directed by the iodo group which subsequently migrates to a position ortho from its initial one. In 1995, as the next stage in this work, we described17c the directed ortho-lithiation of 3-iodo-N,N-diisopropyl-2-pyridinecarboxamide (Scheme 12).
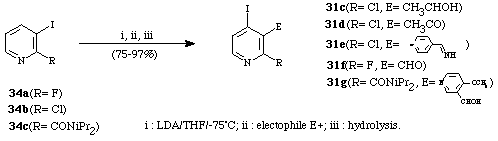
Scheme 12
It should be noted that subsequent oxidation of the alcohol 31g led to the corresponding ketone 31h.
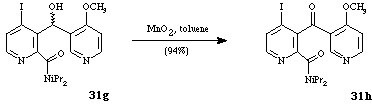
Scheme 13
In the next step of the synthesis (4-pyridyl)borane (31b) as well as 4-iodopyridines (31c-h) were used for the synthesis of benzo[c]-2,7-naphthyridines.
On one hand, palladium catalysed cross-coupling of (4-pyridyl)borane (31b) with ortho-iodoaniline (35) afforded the 4-phenylpyridine 36 which was cyclized under basic conditions to give raw 4-methoxy-5H-benzo[c]-2,7-naphthyridone which was transformed in a triflate derivative 32a. Coupling of 32a with (4-pyridyl)tributylstannane afforded, after cleavage of the ether, the pyridylbenzonaphthyridinone 32b which is an intermediate of the synthesis of amphimedine (28) by Prager19 via a totally different route (Scheme 14).17a
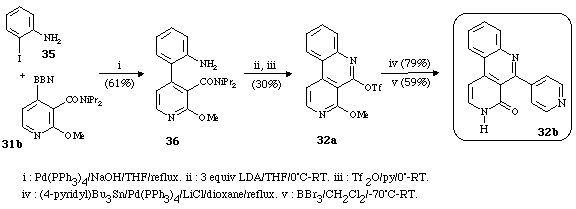
Scheme 14
On the other hand, palladium-catalysed cross-coupling of 4-iodopyridines 31d-f,h with ortho-pivaloylaminophenylboronic acid (37) under Suzuki's conditions afforded 4,5-disubstituted benzonaphthyridines 32. Cyclization of the intermediary 4-phenylpyridine often but not always, occured spontaneously under the basic conditions of the coupling reaction.
In 1993,15 perlolidine (32c) was synthesized in two steps from 2-fluoro-4-iodo-3-pyridinecarboxaldehyde (31f) using a cross-coupling reaction followed by an acidic treatment (Scheme 15).
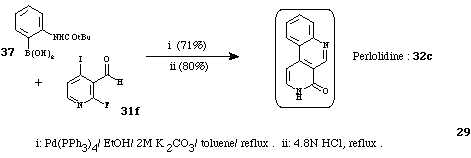
Scheme 15
It could be noted that 2,10-diazaphenanthrene structures 40 were prepared15,18 in a similar way, using a cross-coupling reaction of the pyridine 38 and the phenylboronic acid 39, followed by a treatment with a strong base (Scheme 16).
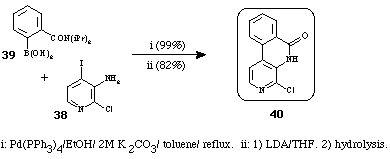
Scheme 16
When reacted with the phenylboronic acid (37) under Suzuki's conditions 31h did not cyclize spontaneously giving the 4-phenylpyridine (41). Cyclization occurs by treatment of 41 with formic acid affording the benzonaphthyridine (32d). This compound could be an intermediate in a new route to meridine alkaloid (30) (Scheme 17).17c
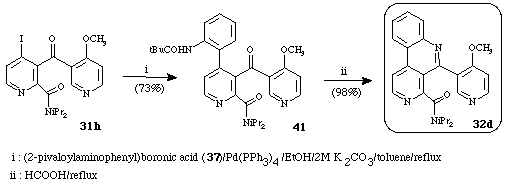
Scheme 17
Cross-coupling of compound 31e, with the phenylboronic acid (37) afforded the 4-chloro-5-(4-pyridyl)-benzo[c]-2,7-naphthyridine (32e) in good yield. Compound 32e is another intermediate in the synthesis of amphimedine by Prager19 (Scheme 17). We found that the overall yield (57% from 34b) could be greatly improved if the reactions were carried out by a one-pot procedure (yield : 82% from 34b)17a (Scheme 17).
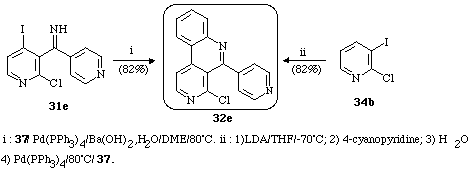
Scheme 18
Finally, we also reported18 in 1995, a new synthesis of 4,5-disubstituted benzo[c]-2,7-naphthyridines (42) based on nucleophilic addition of alkyl- or aryl-lithio derivatives to the corresponding 1-substituted compounds (32f-h) followed by oxidation (Scheme 19).
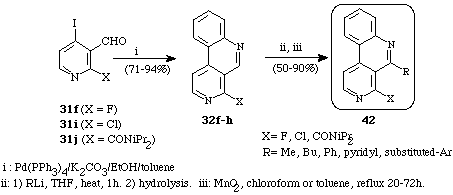
Scheme 19
The high reactivity of the imine moiety allows a full regioselectivity of the
addition and very good overall yields. Some of the resulting 5-aryl compounds
are potential intermediates for marine alkaloids.
Conclusion
Through numerous papers, we have demonstrated that connection between
transition metal catalysed reaction and metalation has become a very efficient tool
for the synthesis of aza-carbazole and benzonaphthyridine structures. Most of
the prepared compounds are natural and biologically active molecules.