| [Molecules: 3] [Related articles/posters: 043 065 071 062 050 ] |
Rearrangement of an epoxide to a carbonyl compound can be catalysed by proton or Lewis acids.[1] In this rearrangement a hydrogen, alkyl or aryl atom migrates between adjacent carbons in the carbonyl-forming step. Many arene oxides react in aqueous solution by both spontaneous and acid-catalysed pathways to give diols and phenols.[2] In this reaction the 1,2-hydride migration between adjacent carbons leading to an intermediate cyclohexadienone is referred to as an "NIH-shift"[3] and occurs in nature, for example, in the biosynthesis of tyrosine.[4] Oxiranes sufficiently activated by substituent aryl and vinyl groups also yield carbonyl products in aqueous solution by the spontaneous reaction pathway.
Acid-induced rearrangement of epoxides by alkyl, aryl or hydrogen migration was for many years considered to be a concerted process. However, for epimeric pairs of exocyclic tertiary substituted epoxides, acid catalysed rearrangement resulted in similar mixtures of epimeric aldehydes.[5] This is schematically represented in Figure 1 and is consistent with a stepwise rearrangement and the intermediacy of carbocations.
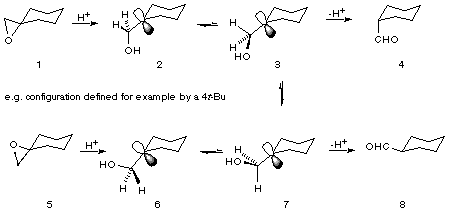
Figure 1 Schematic representation of epoxide rearrangement.
For each epimeric epoxide, where conformation is defined, for example, by a 4tert-butyl group or by fusion to a second ring, a marginal preference for the aldehyde resulting from inversion is observed i.e. 4 from 1 and 8 from 5. The formation, however, of aldehyde 4 along with 8 from 5 is evidence for the intermediacy of a carbocation. A concerted hydride migration from 5 would give only 8. The fact that 4 is also formed is consistent with the reaction proceeding at least in part via a carbocation. On cleavage of the more substituted C-O bond, rotation about the C+-CH2O bond in either direction places a hydrogen anti to the original C-O in a geometry favourable for hydrogen migration. This necessarily occurs before further rotation places a hydrogen syn to the original C-O bond. For example epoxide 5 opens to 6 and rotation to 7 (or the mirror image) occurs before formation of 3 (or the mirror image). Provided that the rate of bond rotation converting 7 to 3 is not rapid relative to hydride migration a preference for formation of 8 from 5 will be observed. By the same argument, reaction of 1 would be expected to give a higher yield of 4 than 8. The extent of bias for the product of inversion at C2 contains information about the relative rate of rotation and hydride migration. For example, if rotation is fast relative to hydride migration then both epimeric epoxides should give an identical ratio of epimeric aldehydes, which is not observed.[6]
Ab initio calculations of the acid-catalysed rearrangement of ethylene oxide to acetaldehyde in the gas phase show protonated epoxide 9 to be an energy minima on the potential energy surface.[7] These early studies of the C2H5O+ potential energy surface at the MP3/6-31G**//RHF/4-31G level suggested carbocation 11 to be an intermediate to protonated acetaldehyde 13. A transition structure 10 between 9 and 11 was identified along with a small activation barrier from 11 to a transition structure for rearrangement to 13. With the inclusion of polarization functions in the basis set and incorporation of electron correlation the energy of 12 falls below the intermediate 11. Thus although cation 11 is predicted to be stable with respect to cyclisation to 9 and with respect to a 1,2-proton shift to protonated vinyl alcohol Radom et al.7 suggested it was unlikely to be an observable species because of facile rearrangement by means of a 1,2-hydride shift to 13.
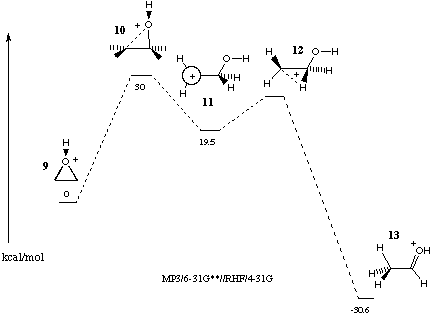
Figure 2 Acid catalysed rearrangement of oxirane
Later calculations at the MP2/6-31G**//HF/6-31G*[8] and MP2/6-31G**//MP2/6-31G*[9] levels predict gas-phase unimolecular ring opening of protonated oxirane 9 to lead to protonated acetaldehyde 13 via an activation barrier of 24.6 and 27.7 kcal mol-1 respectively with no intervening minima. A transition state 11 was established as the C2 cation with the p-orbital orthogonal to the C2C10 plane. The imaginary vibration exhibited a twist of the C2H2 and movement of a C1H into the plane of the p-orbital.8 The migrating hydrogens of 9 in the rearrangement to 13 are diastereotopic with respect to the proton on oxygen. However, the symmetry of the cation 11 precludes differentiation in hydrogens in the migration.
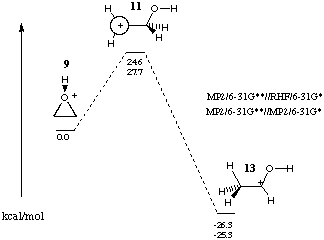
Figure 3 Rearrangement of protonated ethylene oxide.
In contrast, rearrangement of protonated fluorooxirane was calculated (MP2/6-31G*//MP2/6-31G*)9 to open to protonated aldehyde by two stereospecific pathways involving the carbocation intermediates shown in Figure 4. The plane of symmetry of the intermediate carbocations negates diastereoselective selection of a hydride in the migration to protonated aldehyde.
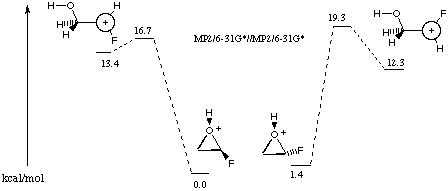
Figure 4 Rearrangement of fluorooxirane
In contrast to the calculations for acid catalysed rearrangement of ethylene oxide, acid-catalysed opening of benzene oxide, styrene oxide and analogous substituted epoxides in the gas phase are calculated by ab initio methods to give carbocations as energy minima.[10] The rearrangement of these epoxides is therefore considered stepwise. For the rearrangement of benzene oxide (14) to phenol, calculations [11] at the MP2/6-31G*//RHF/6-31G* level of theory show the protonated dienone 16 to be an intermediate 13 kcal mol-1 lower in energy than the protonated oxonium ion 15. The protonated dienone 17 resulting from ring opening of 15 followed by an NIH shift is calculated to be 42 kcal mol-1 lower in energy than 15. The oxonium ion 15 was considered to be the transition state for the interconversion of the two otherwise identical carbocation structures in which the HO group is located at adjacent carbons. Carbocation 16 could collapse to phenol by proton loss from C1 or undergo an NIH shift, the latter being favoured (Figures 5 and 6). From studies of deuterium-labelled substrates, it was shown that the NIH pathway is energetically favoured.
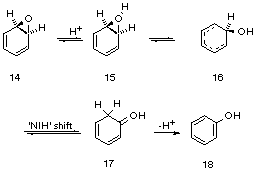
Figure 5 Arene oxide rearrangement
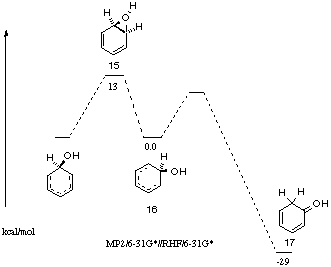
Figure 6 Energy surface for arene oxide rearrangement
Carbocations rather than oxonium ions have been similarly calculated to be intermediates in the rearrangement of monohydroxy, amino and vinyl oxiranes at the RHF/6-31G*//RHF/6-31G* level of theory.10 For oxirane and the fluoro, methyl, cyano, formyl and formaldimino derivatives, both anti and the syn oxonium ion invertomers, which differ in energy by less than 2 kcal mol-1, are calculated to be more stable than the carbonium ions. The corresponding carbocations are intermediates but are consistently higher in energy than the protonated epoxides.
We now report calculations on the proton catalysed rearrangement of propene oxide in order to define in detail the potential energy surface for the more important regions of the rearrangement process.
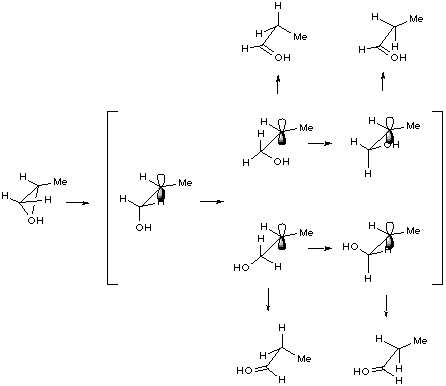
Figure 7 Schematic representation of acid-catalysed rearrangement of propene oxide
Protonation of propene oxide can occur on either face of the oxirane ring, to give the stereoisomeric syn and anti oxonium ions 19 and 21. Such species have been referred to as invertomers.10 At all levels of theory the syn co-ordinated oxonium ion 19 was found to be marginally higher in energy than the trans oxonium ion 21 - at the MP2/6-31G* level of theory by 0.19 kcal mol-1.[14] Protonation of propene oxide is expected to be reversible and the diastereomers most likely interconvert in this way. However an intramolecular transition state 20 has been established 16.85 kcal mol-1 higher in energy to 21.
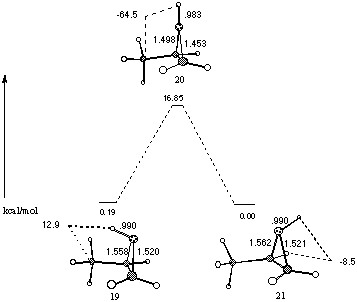
Figure 8 Intramolecular interconversion of stereoisomeric protonated propene oxides
Four transition states involving ring cleavage of 19 and 21 have been established each involving rupture of the more substituted C-O oxirane bond and rotation about C+-CH2O. The energies of these transition states are within 3 kcal mol-1. Importantly the calculations quantify the preference for oxygen to rotate away from the methyl and relieve the skew oxabutane interaction. Such rotation is favoured by ca. 2 kcal mol-1 (Figure 9).[15]
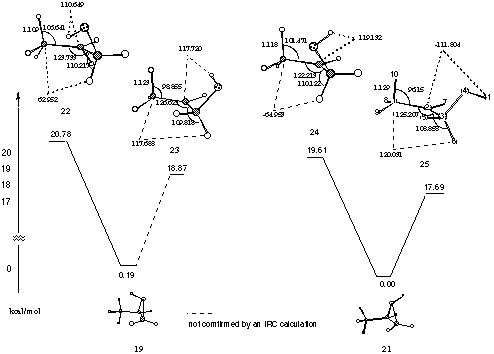
Figure 9 Transition states involved in the potential energy surface for ring cleavage of protonated propene oxide
Three features of the structures are of particular note. The first, the importance at the transition state of CH hyperconjugation from the methyl hydrogen in the plane of the carbocation p-orbital. This CH is bent towards the carbocation and the H10C1C2 angle in 25 is 96o. Furthermore the CH bond is extended in length (1.129 Å). Similar but less marked effects are observed in the other three higher energy transition states. The second feature of note is that at the transition states the epoxide has opened so that C2 and C3 are essentially sp2 and sp3 hybridised respectively - the C2C3H6 angles are 109-110o and the C1C2C3 angles 122-126o. Finally of note is that C6H has not started to migrate at the transition states nor is the C6H bond aligned with the carbocation p-orbital - the C8C2C3H6 dihedral angles being some 25 to 30o from the plane of the carbocation p-orbital.
In view of the large structural differences between transition structures and reactants and products, IRC paths were determined to establish connectivities. The most favoured pathway for ring cleavage involves transition state 25. It seemed pertinent to study this pathway in some detail. An intrinsic reaction coordinate calculation (IRC) was performed at the MP2/6-31G* level and showed the transition state to collapse to protonated epoxide 21 and protonated aldehyde in conformation 11 (Figure 10).
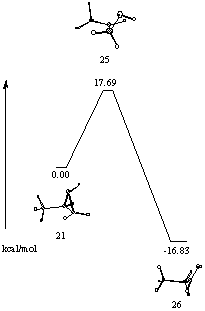
Figure 10 Intrinsic reaction coordinate relating 21 and 26
Analysis of the reaction trajectory reveals two distinct and interdependent processes. The first, rupture of the oxirane and the second hydride migration. The overall reaction profile via this transition state is consistent with an asynchronous concerted rearrangement elaborated in some detail below.
The variation of bond length and angle with reaction coordinate, shown in Figure 11, reflects the importance of hyperconjugation of a methyl hydrogen with the carbocation centre - H10C1 is longest, H10C1C2 most compressed and the dihedral angle H10C1C2C3 reaches a minimum of 96o at the transition state. This demand for hyperconjugation at the transition structure is consistent with the charge development at C2 being greatest at this point in the reaction coordinate.
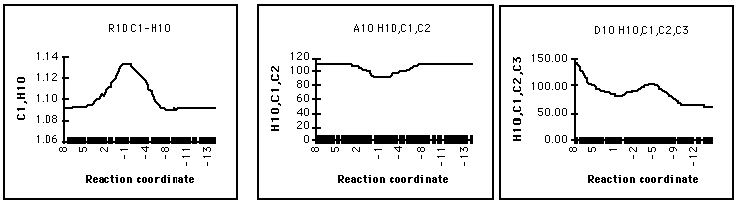
Figure 11 The importance of hyperconjugation
At the transition state the oxirane ring has opened such that C2 and C3 are essentially sp2 and sp3 hybridised respectively. With rupture of the epoxide the OC3C2 angle increases so that at the transition state this angle is 102o just short of tetrahedral and thereafter increases to that for sp2 hybridisation as in protonated aldehyde (Figure 12). Concomitant with C2O bond rupture is rehybridisation at C3 to tetrahedral. A notable feature apparent in the plot of C3O bond distance with reaction coordinate is that the double bond character of the protonated carbonyl is formed late in the reaction coordinate, in a second step of the reaction profile.
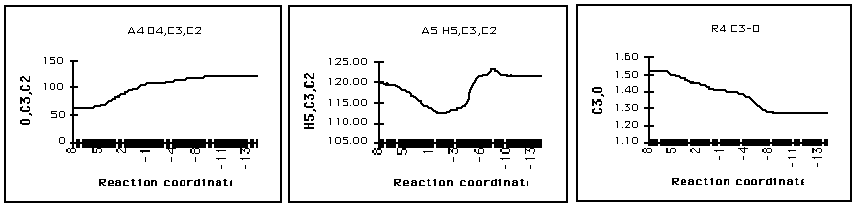
Figure 12 Oxirane ring rupture
The migration of hydride is late in the reaction coordinate with the C3H6 distance remaining constant well past the stage where the oxirane ring has ruptured - the transfer of hydride coinciding with a change in the hybridisation at C2 from sp2 to sp3. This is reflected by the C3C2C1 and H6C3C2 angles (Figure 13).
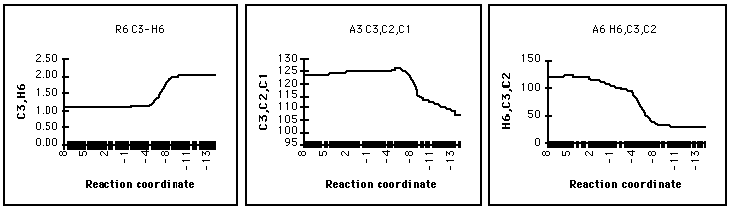
Figure 13 Hydride migration
Two other stationary points 28 and 29 were established on the reaction surface (Figure 14). For both these structures C1C3C3OH are planar. Structure 29 is flanked by minima where the eclipsing of the CO with the adjacent CH is relieved by a change in the torsional angle of 2o. Surprisingly 29 is computed at the MP2/6-31G* level to be higher in energy than 28 where the oxygen is cis to the methyl. These structures are being further investigated. An IRC calculation shows the connection of 19 via transition state 22 to 28. A surface between 28 and aldehyde has not been established. The importance of 29 on the reaction surface was also difficult to elaborate but the structure optimised to 26, shown by a dotted line in Figure 14.
An IRC calculation of transition structure 24 shows it to be linked to aldehyde 27 and protonated epoxide 21. The calculation failed before reaching the aldehyde and protonated epoxide but had progressed sufficiently in each direction to establish this concerted asynchronous pathway between 21 and 27. The broken lines represent extrapolation based on the last geometry reached. At each of the break points in Figure 14, the IRC algorithm suddenly failed to optimize to the IRC path and terminated abruptly with a bizarre extrapolation of the geometry. We speculate that the failure may be due to a bifurcation point, i.e. a point where a zero force constant for a normal mode perpendicular to the IRC is encountered. Conformation 27 of the protonated aldehyde is computed to be 0.5 kcal mol-1 higher in energy that conformation 26 consistent with the known preference of a carbonyl to eclipse with a C-C bond in preference to a C-H bond.
An IRC calculation for transition structure 23 showed it going to 26 but the surface on the other side of the transition state was difficult to define because methyl rotation resulted in a minimum close to transition state 23 despite the fact that atomic trajectories of 23 along the normal mode with imaginary frequency gace no evidence for this motion.
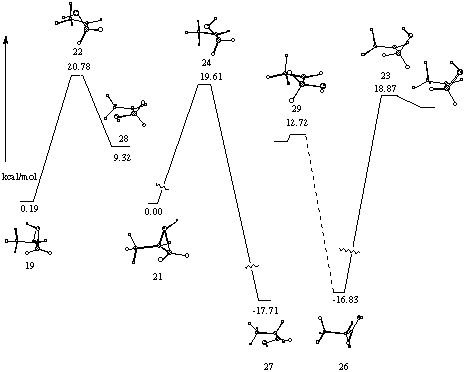
Figure 14 Reaction surface connecting protonated propene oxides and aldehydes
The lowest energy concerted reaction coordinate diagram connecting 21 and 26 via transition state 25 summarises an asynchronous but concerted reaction surface for the acid-catalysed rearrangement of propene oxide (Figure 15).
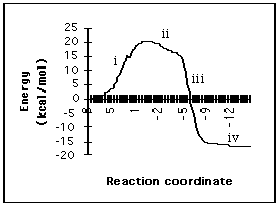
Figure 15 Lowest energy reaction coordinate diagram connecting 21 and 26 via transition state 25. (i) ring opening, (ii) preparation for hydride transfer, (iii) hydride transfer, (vi) approach to a low energy conformation of protonated aldehyde.
The reaction surface is characterised thus (i) ring opening which leads directly to the transition state 25, (ii) preparation for hydride transfer stage (i.e. the plateau), (iii) hydride transfer itself at the rapid drop from the plateau, (iv) a flat approach to a low energy conformation of protonated aldehyde. Unlike a tertiary epoxide (Figure 1) the calculations show a potential energy surface for rearrangement of protonated propene oxide which does not require a discrete carbocation intermediate. Any diastereoselectivity in the rearrangement will reflect the relative preference for the transition states 25 and 23 versus 24 and 22 (Figure 9).
References
1 Norman, R.O.C.; Coxon, J. M. Principles of Organic Synthesis, 3rd Ed.,
Blackie, Chapman and Hall, London, 1993, p. 590.
2 Islam, N. B.; Gupta, S. C.; Yagi, H.; Jerina, D. M.; Whalen, D. L. J. Am. Chem. Soc. 1990, 112, 6363. Nashaat, N. T.; Balani, S. K.; Loncharich, R. J.; Shipley, D. Y.; Mohan, R. S.; Whalen, D. L.; Jerina, D. M. J. Am. Chem. Soc. 1991, 113, 3910.
3 Jerina, D.; Daly, J.; Witkop, B.; Zaltaman-Nirenberg, P.; Underfriend, S. Arch. Biochem. Biophys. 1969, 128, 176. Jerina, D. M.; Daly, J. W.; Witkop, B.; Zaltaman-Nirenberg, P.; Underfriend, S. Biochemistry, 1970, 9, 147. Jerina, D. M.; Daly, J. W.; Witkop, B.; Zaltaman-Nirenberg, P.; Underfriend, S. J. Am. Chem. Soc. 1968, 90, 6523, 6525. Boyd, D. R.; Jerina, D. M.; Daly, J. J. Org. Chem. 1970, 35, 3170. Boyland, E.; Sims, P. Biochem. J. 1965, 95, 788.
4 Dipple, A.; Moschel, R. C.; Bigger, A. H. Chemical Carcinogens, 2nd edn., Searle, C. E., Ed., Vol. 1, ACS Monograph, 182, American Chemical Society, Washington, DC, 1984, p. 41. Polycyclic Hydrocarbons and Carcinogenesis, Harvey, R. G., Ed., ACS Symposium Series 283, American Chemical Society, Washington, DC, 1985. Polycyclic Hydrocarbons and Cancer, Gelboin, H. V.; Tso, P. O. P., Eds.; Vol. 1, Academic Press: New York, 1978.
5 Blackett,B.N.; Coxon,J.M.;Hartshorn, M.P.; Jackson, B.L.J.; Muir, C.N. Tetrahedron, 1969, 25, 1479.
6 Blackett, B. N.; Coxon, J. M.; Hartshorn, M. P.; Richards. K. E. Aust. J. Chem. 1970, 23, 839. Blackett, B. N.; Coxon, J. M.; Hartshorn, M. P.; Richards, K. E. J. Am. Chem. Soc. 1970, 92, 2574. Blackett, B. N.; Coxon, J. M.; Hartshorn, M. P. Richards, K. E. Aust. J. Chem. 1970, 23, 2077. Coxon, J. M.; Chom-Ey Lim. Aust. J. Chem. 1977, 30, 1137. Coxon, J. M.; McDonald, D. Q. Tetrahedron Lett. 1988, 29, 2575.
7 Nobes, R.H.; Rodwell, W.R.; Bouma, W.J.; Radom, L. J. Am. Chem. Soc. 1981, 103, 1913.
8 Ford, G. P.; Smith, C. T. J. Am. Chem. Soc. 1987, 109, 1325
9 Bock, C. W.; George, P.; Glusker, J. P. J. Org. Chem. 1993, 58, 5816.
10 George, P.; Bock, C.W.; Glusker, J.P. J. Am. Chem. Soc. 1992, 96 7302
11 George, P.; Bock, C.W.; Glusker, J.P. J. Phys. Chem. 1990, 94, 8161
12 GAUSSIAN94, Revision B.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1995.
13 Curtiss, L.A.; Raghavachari, K. Pople, J.A. J. Chem. Phys. 1993, 98, 1293.
14 Similar results have been noted by George11 who reported that the inclusion of electron correlation at the MP2/6-31G*//RHF/6-31G* level does not change these small energy differences to any appreciable extent.
15 The energy difference is greater than between 19 and 21 which reflects the hydrogen atom configuration in the two diastereomers.