Molecular mechanics calculations
The MM+ force field is an extension of MM2 which was developed by Allinger and coworkers.
The conformational analysis of the diastereomers 2a,2b,4a and 4b was performed by using this force field and the Pollack-Ribiere optimizer. The electrostatic interactions were calculated using the bond dipoles included in the MM+ parameters set.
The minimum energy conformations for compounds 2a and 2b are shown below.
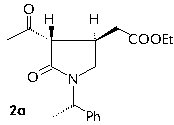


 The energy difference beetween 2a and 2b is 1.21 kcal mol-1 (1 cal = 4.184 J) being 2a more stable than 2b. This energy difference is probably due to a particularly stabilizing pi-stacking interaction between the phenyl of the chiral group and the COOEt group which lie at the same side in the low energy conformation for 2a.
The energy difference beetween 2a and 2b is 1.21 kcal mol-1 (1 cal = 4.184 J) being 2a more stable than 2b. This energy difference is probably due to a particularly stabilizing pi-stacking interaction between the phenyl of the chiral group and the COOEt group which lie at the same side in the low energy conformation for 2a.
MM+ supplements the standard MM2 force field by providing additional parameters.
As references see: (a) Allinger, N.L., J. Am. Chem. Soc., 1977, 99, 8127. (b) Allinger, N.L., Yuh, Y.H., Quantum Chemistry Program Exchange, Bloomington, Indiana, Program No. 395, Molecular Mechanics, Burkert, U.; Allinger, N.L., ed., ACS Monograph 177, American Chemical Society, Washington, D.C., 1982. (c) Lii, J.; Gallion, S.; Bender, C.; Wikstrom, H.; Allinger, N.L., Flurchick, K.M.; Teeter, M.M., J. Comp. Chem., 1989, 10, 503. (d) Lipkowitz, K.B., QCPE Bulletin, Indiana University, 12, 1 (Feb. 1992). The program is enclosed in Hyperchem package, available from Hypercube, Waterloo, Canada.
As reference see: G.B.Jones, B.J.Chapman, Synthesis, 1995, 475.
 Return to poster main page
Return to poster main page