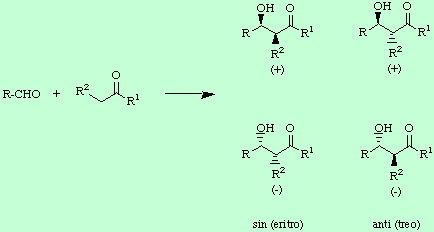
En este sentido hay dos aspectos importantes a considerar:
a) El llamado control estereoquímico interno o diastereoselección, que se refiere a la producción desigual de un par de enantiómeros, ya sea el sin o el anti, sobre el otro. Por tanto, hablar de diastereoselección significa hablar del control de la estereoquímica relativa C-2/C-3 sin o anti.
b) El control estereoquímico absoluto o enantioselección, que favorecerá la producción desigual, en un par determinado, de un enantiomero sobre el otro, ya sea el (+) o el (-).
En cuanto al primer punto, es decir la diastereoselección, se puede decir que depende de las condiciones de reacción empleadas, ya sean éstas de control cinético o termodinámico. Bajo condiciones de control cinético, como se verá más adelante, la producción diastereoselectiva de un par sin o anti vendrá controlada por la geometría del enolato. Si dicha geometría es cis se obtendrá la pareja de aldoles sin y si por el contrario el enolato empleado es trans el producto aldólico será el par anti.
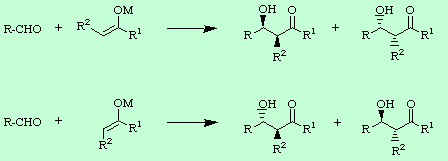
Sin embargo, bajo condiciones de control termodinámico siempre se obtendrá el producto anti, con independencia de la geometría del enolato, ya que la reacción de adición aldólica es reversible con posibilidad de establecer un equilibrio sin/anti, que va siendo desplazado hacia la formación del producto anti, termodinámicamente más estable.
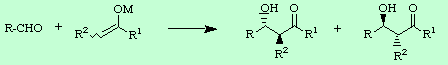
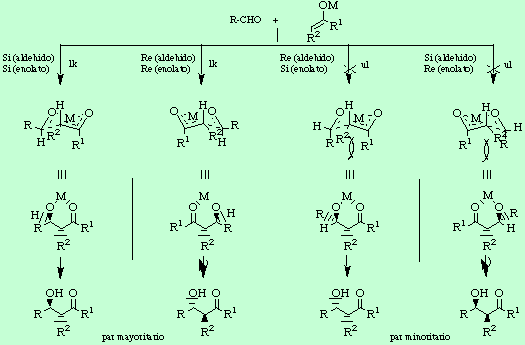
La diastereoselección sin/anti bajo condiciones de control cinético se podría haber predicho en base a un estado de transición cíclico de seis eslabones, tipo silla, conocido como modelo de Zimmermann-Traxler[1]. Por ejemplo, para la combinación de un aldehído con un enolato de geometría cis resultan cuatro posibilidades, con las aproximaciones de aldehído y enolato representadas en el siguiente esquema (en todas ellas el átomo metálico está coordinado a los átomos de oxígeno carbonílico y del enolato). Sin embargo, los dos últimos estados de transición, a diferencia de los dos primeros, son altamente desfavorables debido a las interacciones 1,3-diaxiales entre los restos alquilo R y R1. Por tanto, una de las condiciones a la hora de aplicar este modelo de Zimmermann-Traxler es situar el resto R del aldehído en una posición ecuatorial. Centrándonos ahora en los dos estados de transición enantioméricos resultado de las dos posibles combinaciones ul y trasladándolos a formas en zig-zag, se puede visualizar la disposición sin de los grupos hidroxilo y alquilo en los estereocentros creados.
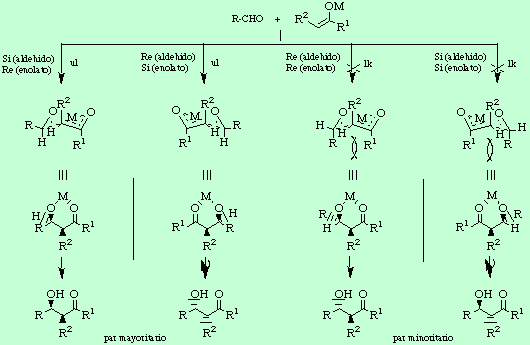
Como prueba experimental de la validez de este modelo cabe decir que conforme aumenta el volumen de los sustituyentes R sobre el aldehído y R1 sobre el enolato la diastereoselectividad sin es tanto mayor.
Si el mismo tratamiento se realiza para un enolato trans y se razona en los mismos términos que en el caso anterior, es decir estados de transición desfavorecidos aquellos en los que hay interacciones 1,3-diaxiales entre restos alquilo y favorecidos aquellos en los que el grupo R del aldehído adopta una posición ecuatorial, se puede entender por qué ahora se obtiene mayoritariamente el par anti de productos aldólicos.
![]()
![]()
![]()