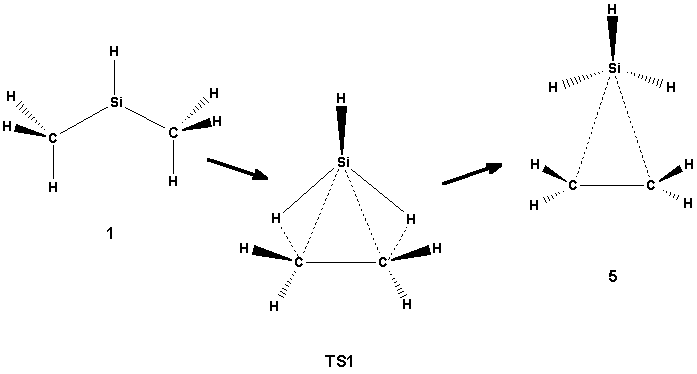
Scheme 1
The ion structures were fully optimized at different levels of theory, starting from the SCF level, where three different basis sets were employed: 3-21G, 6-31G* and 6-31G**. In addition, calculations were also performed using second-order many-body perturbation theory (MP2) and a hybrid DFT-HF method using a combination of Becke's three-parameter exchange functional and the Lee-Yang-Parr non-local correlation functional (B3LYP). Zero-point vibrational energy corrections (ZPVE) were only introduced at the B3LYP level, since this method gives frequencies in close agreement with experiment.
Five isomers of the SiC2H7+ ion have been found. The first one, which may be described as (CH3)2SiH+ (1, Scheme 1), is obviously the global minimum, although it was not considered in the work by Apeloig et al. [2] , the most elaborate study of this ion that we have found. According to their work, the most stable species is C2H5SiH2+ (2, Scheme 3), and the silyl-bridged H3SiCH2CH2+ (5, Scheme 2), was found to lie only 5.5 kcal/mol higher in energy. Apeloig et al. discuss also MeSiH3CH+ (4 in our notation, Scheme 2), which according to their calculation lies 31 kcal/mol higher in energy than their most stable isomer. In addition, we have also studied CH3SiH2CH2+ (3a, Scheme 4).
Scheme 1
All these five isomers have been fully optimized at different levels of theory, For the isomers 1 and 2 (which were found to have Cs symmetry) there were no dramatic changes in geometry or energy in going from SCF to correlated levels of theory. Isomer 5 was also found to have a Cs structure with the SiH3 group located symmetrically at the plane bisecting the ethene molecule (Scheme 1). This structure may therefore be regarded as a complex between the SiH3 cation and the ethene molecule.
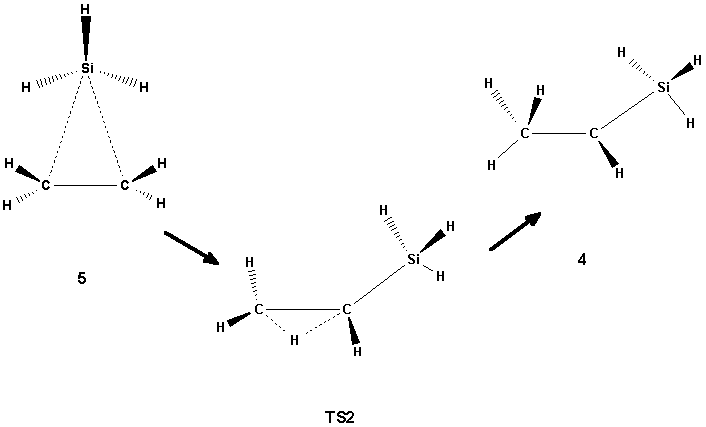
Scheme 2
The most dramatic changes occurred for the isomers 3 and 4 when going from SCF to correlated methods. The SCF calculations give a structure which can be described by CH3SiH2CH2+ (3a), but the MP2 calculation gives an increased Si-C bond length and decreased C-Si-C bond angle (3b, scheme 4), whereas the B3LYP calculation results in a shift of the methyl group towards the other carbon atom, giving the 2 structure, meaning there is no minimum for the 3a isomer at this level of theory.
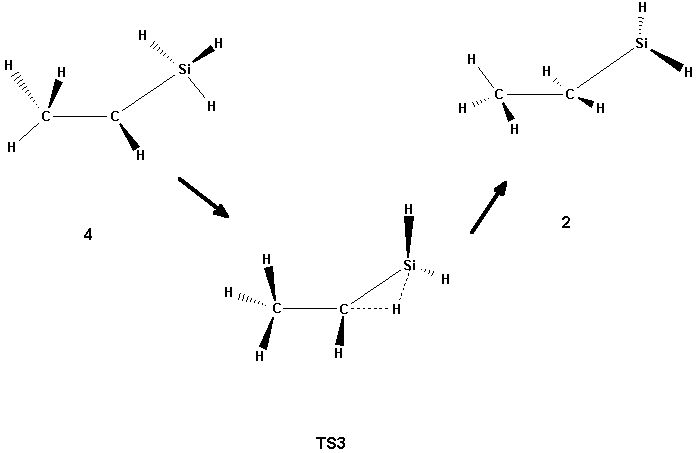
Scheme 3
The isomer 4, on the other hand, has a B3LYP structure which is similar to that found at the SCF level, but at the MP2 level its structure is characterized by the hydrogen atom being in the bridging position between the two carbon atoms. The structure 4 may be regarded as an intermediate between 2 and 5, which can be produced from 4 by hydrogen shifts. Two transition states have been found for these reactions: TS2 which connects 4 and 5 (Scheme 2), and TS3 which connects 4 and 2 (Scheme 3) by shifting the silyl group towards the central carbon. No transition state for the direct interconversion between 1 and 2 was found. For the transition between 1 and 5 only one transition state (TS1, Scheme 1) was found. The TS1 structure is characterized by the symmetric shift of the three hydrogens of the silyl group of 5 towards the other carbons. In contrast to TS2 and TS3, where there are no dramatic changes in the energies when going from SCF to correlated methods, the height of the TS1 barrier decreases significantly when the correlation is switched on (cf. Table 1).
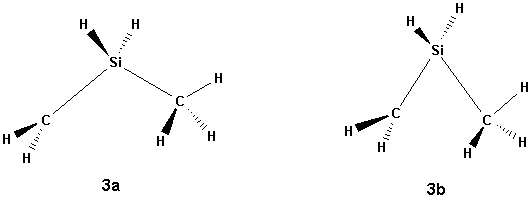
Scheme 4
The relative energies of the five different isomers and the three transition states are shown in Table 1. As is shown by the Table, the complexation energy of C2H4 + SiH3+ is sufficient to drive the conversion between isomers, which has been observed experimentally for the silylenium cation [3,4].
| Stationary points | SCF | MP2 | B3LYP | |||
|---|---|---|---|---|---|---|
| 3-21G | 6-31G* | 6-31G** | Ee | E0* | ||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 24.7 | 20.1 | 20.9 | 21.0 | 22.7 | 22.7 |
| 3 | 54.2 | 47.1 | 47.6 | 18.2 | - | - |
| 4 | 50.9 | 38.5 | 39.0 | 47.0 | 44.4 | 41.1 |
| 5 | 35.0 | 23.3 | 23.9 | 25.5 | 25.4 | 24.3 |
| TS1 | 97.6 | 87.8 | 86.7 | 67.8 | 65.5 | 63.2 |
| TS2 | 61.8 | 43.5 | 43.0 | 49.4 | 47.3 | 43.9 |
| TS3 | 60.8 | 45.6 | 45.8 | 52.8 | 45.5 | 42.3 |
| C2H4+SiH3+ | 75.3 | 65.8 | 65.6 | 72.9 | 74.2 | 69.6 |
*E0=Ee+ZPVE
2. Apeloig, Y.; Karni, M.; Stanger, A.; Schwarz, H.; Drewello, T.; Czekay G. J. Chem. Soc., Chem. Commun. 1987, 989.
3. Bakhtiar, R.; Holznagel,C.M.; Jacobson, D.B. Organometallics 1993, 12, 621.
4. Bakhtiar, R.; Holznagel,C.M.; Jacobson, D.B. Organometallics 1993, 12, 880.