A Detailed Description of Conformational Space of Native
Enkephalines and their Cyclic Analogues
(a)
Laboratory of Computational Chemistry and Molecular Modeling,
Department of Organic Chemistry, Masaryk University, 611 37 Brno, Czech
Republic
(b) Institute of Organic Chemistry, University of Trondheim, Norwegian
Institute of Technology, N-7034 Trondheim-NTH, Norway
Introduction
The discovery of two endogenous pentapeptides with morphine-like
action, Met-enkephalin (Tyr-Gly-Gly-Phe-Met) and Leu-enkephalin
(Tyr-Gly-Gly-Phe-Leu), have induced considerable interest as to the
activities and conformational properties of opioid peptides, which are
of interest as possible substitutes for alkaloid opiate drugs and for
their biological importance as endogenous analgesies. Because of their
varied and important biological roles, the structural features of enkephalines
and their analogues have been extensively investigated over the past 20
years by various methods including spectroscopy (NMR, CD, IR, UV), X-ray
crystallography and molecular modeling [1]. These linear small flexible molecules
that, due largely to this flexibility, can interact with several distinct
receptor types, each requiring different ligand conformations, to initiate
different physiological events. One method reducing the flexibility of these
peptides is cyclization. This not only greatly reduces the flexibility,
but has also led to the synthesis of many active and receptor selective
analogues. For our study we have chosen:
1. linear enkephalines
- Leu-enkephalin (Tyr-Gly-Gly-Phe-Leu)
|
 |
- Met-enkephalin (Tyr-Gly-Gly-Phe-Met)
|
 |
2. cyclic analogues of enkephalines
- DLFE (Tyr-c(D-Lys-Gly-Phe))
|
 |
- DPDPE (Tyr-c(D-Pen-Gly-Phe-D-Pen))
|
| | |
Methodology
We have performed a detailed conformational study using the ROSE[2]
, CICADA[3]
and PANIC[4] software. For energy computation we have used the
PMMX program, which is the MMX[5] program specially parametrized for
peptides[6]. The interface between the programs is schematically illustrated
in the followed scheme.
Scheme 1
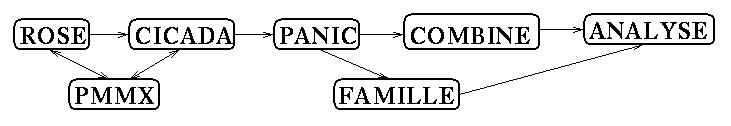
Program ROSE is used for generation of the starting conformations by
a combinatorial algorithm. Program CICADA performs travelling along the PES.
Energy of the structures are computed by program PMMX which is MMX program
specially parametrized for peptides. Results from the program CICADA are used
for PES and flexibility analysis which is performed by program PANIC. Results
from the programs CICADA and PANIC serve for the simulation of the conformational
travelling along the PES which is performed by program COMBINE. Program ANALYSE
is used to extract geometry parameters of structures obtained by COMBINE
run. The data could be visualized by the graphics interface.
Results
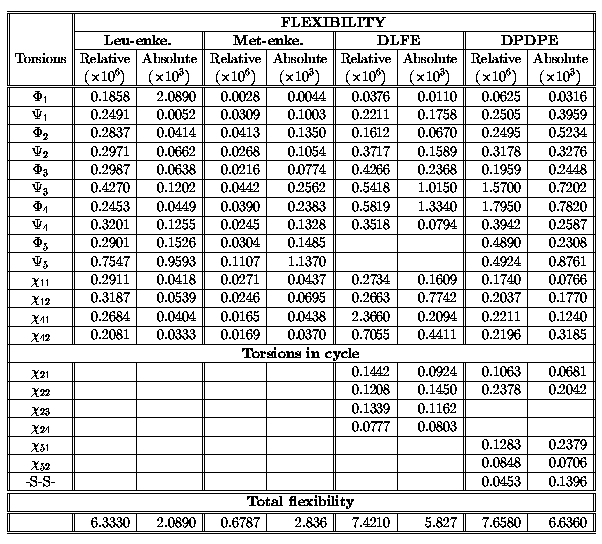
Computed relative and absolute flexibilities[7] of the all investigated
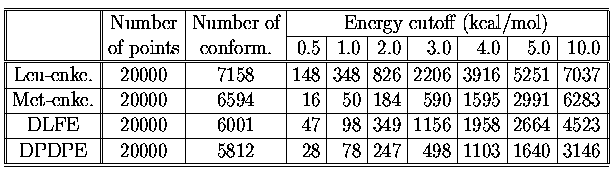
molecules are summarized in Table 1.
Table 1
It is shown, that both relative and absolute flexibilities[7] of linear
enkephalines are comparable with flexibility numbers of cyclic analogues.
Flexibility numbers of the side chain torsions are similar to flexibility
numbers of the backbone torsions, which is different than computed flexibilities
of single amino acids[9]. Torsions of the backbone of the peptides are more rigid
than the same of the amino acids due to the arrangement in peptides. The major
part of flexibility is concentrated on the outside backbone torsions of the leucine
and methionine of the linear enkephalines and on the torsions of the glycine
and phenylalanine of the cyclic enkephalin analogues. The most rigid
from the relative and absolute flexibilities point of view is Met-enkephalin.
Computed flexibilities reflect the numbers of conformers found in various
energy cutoff which are summarized in next the table. The number of points
is the total number of nuclear configurations saved by CICADA travelling
along the PES. In order to allow for comparison of results, the calculation was
stop ed on the same number of points for each molecule.
Table 2
It is shown, that the total numbers of conformations calculated for the same
number of points are comparable for all molecules.
The number of conformers in 10 kcal/mol energy window are for
linear peptides almost two times bigger then for the cyclic analogues.
The most rigid molecule , met-enkephalin,
has the lowest number of conformer in energy windows under 2 kcal/mol.
It is interesting that DPDPE, which is the most flexible molecule from
the flexibility numbers point of view, has the lowest number of conformers
in 10 kcal/mol energy window.
This is in contradiction with
the assumption, that a flexible molecule has a large number of low
energy conformers. Explanation of this phenomenon is shown in the next table.
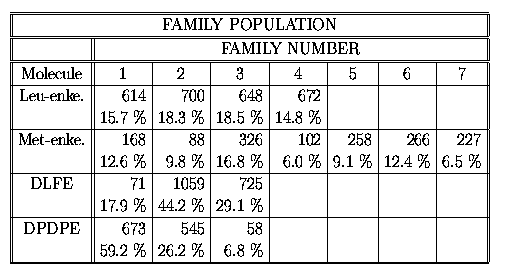
We have performed the clustering of the conformations found into
conformational families[8]. As a criteria of the distribution, we have included
the differences between backbone torsions.
Table 3
It is shown that the cyclic analogues fit just
one very large conformational family with Boltzmann probability around
50 % and two smaller families. The conformations of the linear peptides are
spread into four (leu-enkephalin) and seven (met-enkephalin) families with even
distributed Boltzmann probability around 15 %. The total numbers of
families found are as follows: leu-enkephalin - 335,
met-enkephalin - 382,
DLFE - 44 and DPDPE - 34.
It is shown, that conformations of linear peptides
are distributed in large number of families with large distances and
with large energy barriers separating them, whilst conformations of the cyclic
analogues were found in low number of families with conformations
near by each other and with small interconversion energy barriers. The
computed flexibilities reflect this phenomenon. Our assumption is
confirmed by RMS analysis of the dynamics simulation based on interconversion
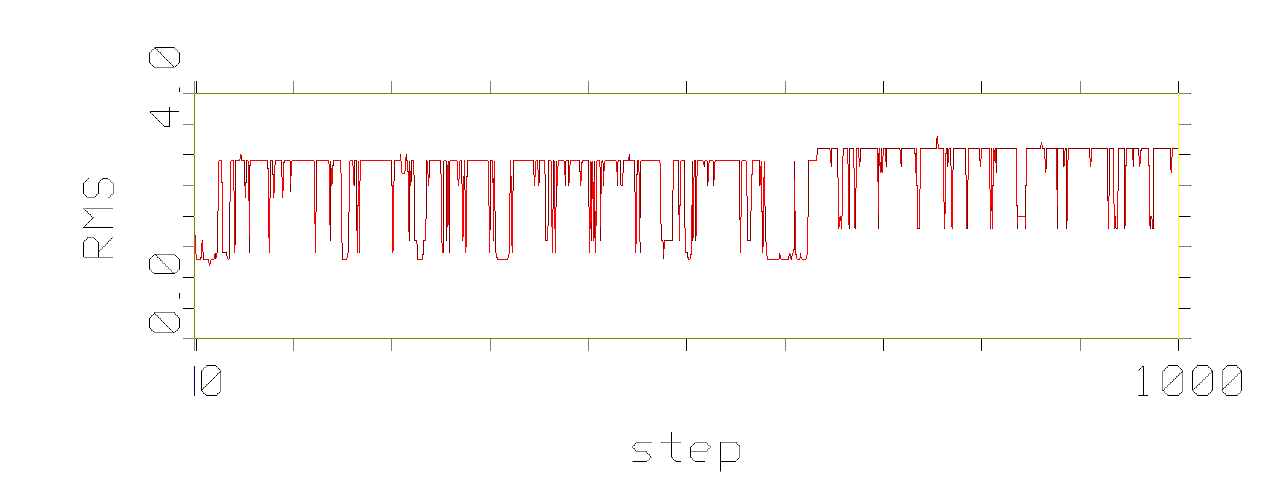
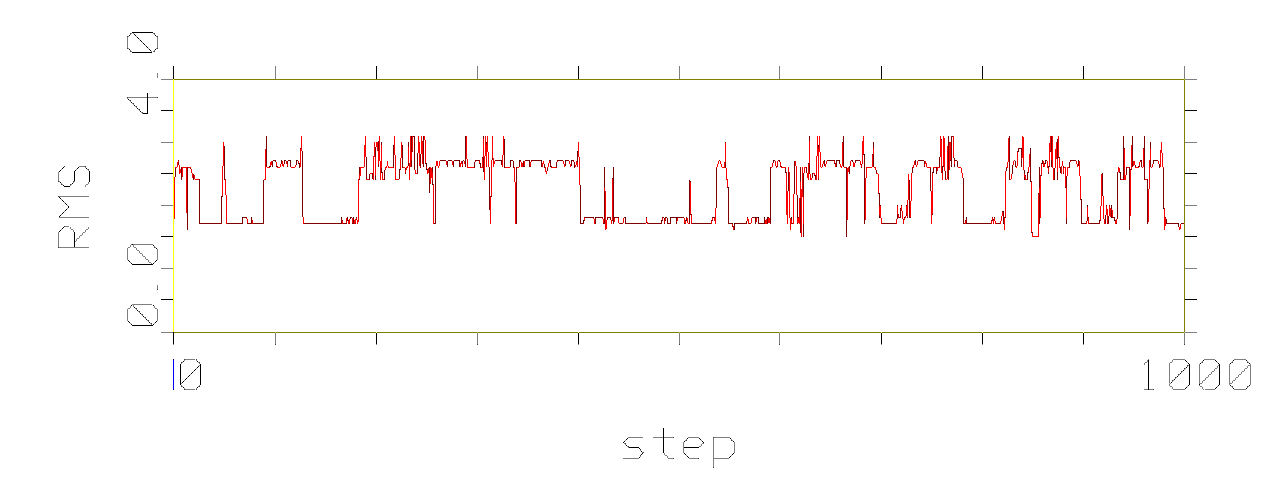
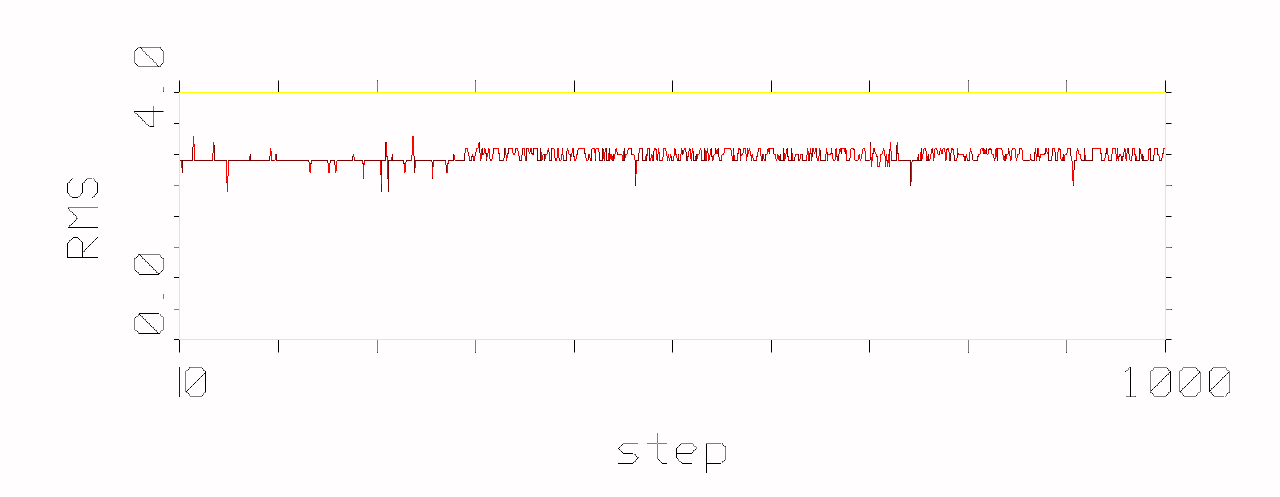
phenomenon, which is pictured in the following figures.
Figures show development of RMS differences during the simulation of
conformational movement within the step window 0-1000 at the thermodynamic
temperature 300 K. All the structures on the trajectory are compared with
the lowest energy conformer.
It has been observed that conformational movement of the linear peptides
is accompanied by much larger RMS differences than that of the cyclic analogues.
Conclusions
Conformational potential energy hypersurface for linear enkephalines and
their cyclic analogues have been analyzed by means of molecular mechanics
in combination with programs ROSE, CICADA, PANIC, COMBINE and ANALYSE.
- Cyclic analogues of enkephalines fit just one highly populated family,
while linear enkephalines are spread into several even populated families.
- The conformational movements of the cyclic analogues are more restricted
than those of the linear enkephalines.
- Cyclic and linear enkephalines are comparable from the flexibility
numbers point of view.
References
- [1] W. H. Graham, E. S. Carter II and R. P. Hics: Biopolymers, 1755-1764,
32, 1992.
- [2] J. Koca, P. H. J. Carlsen: J. Mol. Struct. (Theochem),131, 257, 1992.
- [3] J. Koca: J. Mol. Struct. (Theochem), 13, 308, 1994.
- [4] J. Koca, S. Perez, A. Imberty: J. Comput. Chem., 296, 16, 1995.
- [5] J. J. Gajewski, K. F. Gilbert: MMX is an extended and improved version
of Allinger's MM2. This program is available from Serena software, Box 3076,
Bloomington, IN 47402-3076.
- [6] S. Wolfe, D. F. Weaver, K. Yang: Can. J. Chem. 2687, 66
, 1988.
- [7] J. Koca: J. Mol. Struct., 125, 343, 1995.
- [8] A. Imberty, S. Perez: Glycobiology, 351, 4, 1994.
We thank to Dr. Anne Imberty for providing us with the program.
- [9] J. Koca, Z. Kriz, P. H. J. Carlsen: J. Mol. Struct. (Theochem), 157,
306, 1994.
Acknowledgements
This work has partially been supported by the Grant Agency of the Czech
Republic,
grant No. 203/94/0522, and by Chem-Consult, Hundhamaren, Norway. This financial
support is gratefully acknowledged. We would also like to thank Academic
Supercomputer Center in Brno for providing us access to the computer facilities.
The academic license provided by Biosym/MSI, Inc., for Insight II software is
also acknowledged.
{kind=link}
{kind=link}
{kind=link}
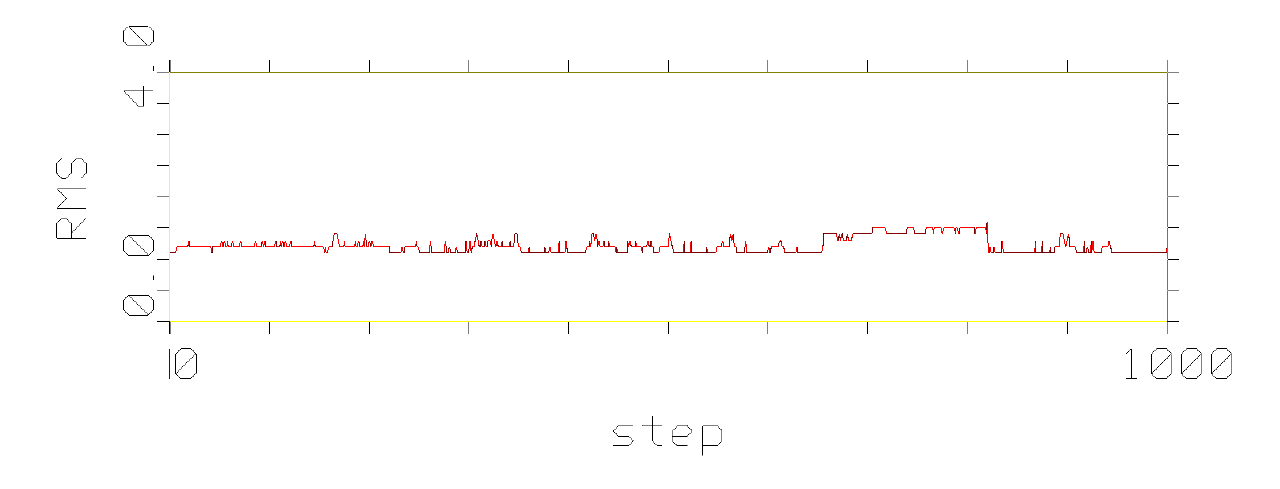{kind=link}