 |
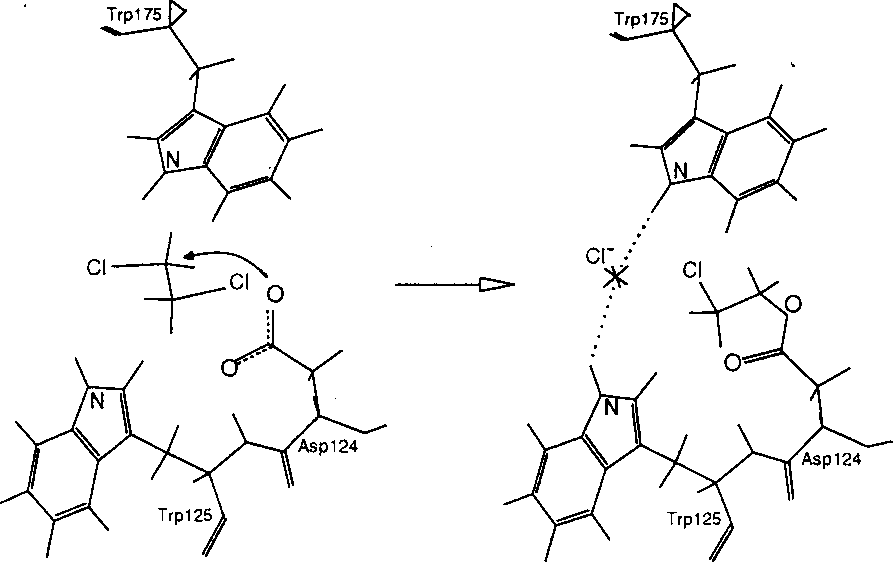
The haloalkane dehalogenase of Xanthobacter autotrophicus GJ10 [1] is an enzyme, which catalyses the environmentally important detoxification process - the hydrolytic cleavage of the carbon-halogen bond. The nucleophilic substitution bimolecular (SN2) reaction mechanism has been proposed for the first reaction step (Fig. 1) based on X-ray crystallography experiments [2]. Here we describe the semi-empirical AM1 study of the kinetic and thermodynamic characteristics of the first reaction step of the dehalogenation reaction.
|
Scheme of the first reaction step catalysed by the haloalkane dehalogenase. The deprotonated oxygen atom of Asp124 performs a nucleophilic attack on the substrate (e.g., 1,2-dichloroethane) carbon atom resulting in the formation of ester intermediate and the release of the chlorine anion.
The cartesian coordinates of the enzyme-substrate complex of Xanthobacter autotrophicus GJ10 haloalkane halidohydrolase soaked with 1,2-dichloroethane were obtained from the Brookhaven Protein Database (2DHC). The subroutine DRIVER [3] of the semi-empirical quantum chemical program MOPAC6.0 [4] was used for the mapping of the reaction pathway of the first step (Fig. 1) of the dehalogenation reaction catalysed by the haloalkane dehalogenase. The distance between the nucleophilic oxygen of aspartic acid (Asp124) and the carbon atom of 1,2-dichloroethane (DCE) was driven - decreased by defined increments (0.05 A or 0.1 A) while optimizing the rest of the structure. The input structures of various complexities were used in the MOPAC/DRIVER calculations (Fig 2a-c). The structures of the single mutant enzymes (Table 1) were modelled and optimised using molecular mechanic method (cvff forcefield of DISCOVER - Biosym program). All the drawings present were prepared using InsightII Biosym software.
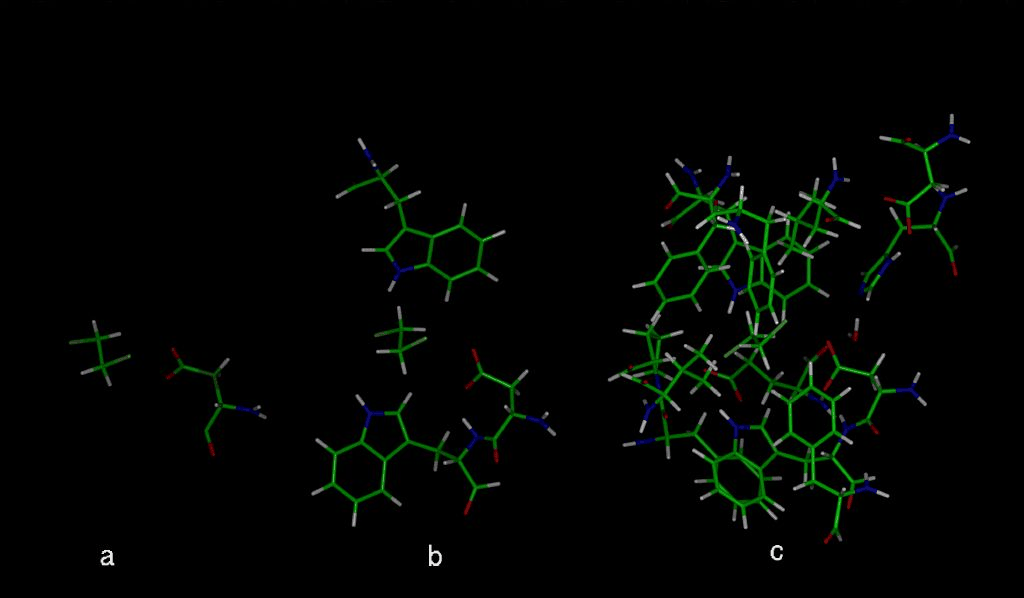 |
The input structures used in MOPAC/DRIVER calculations.
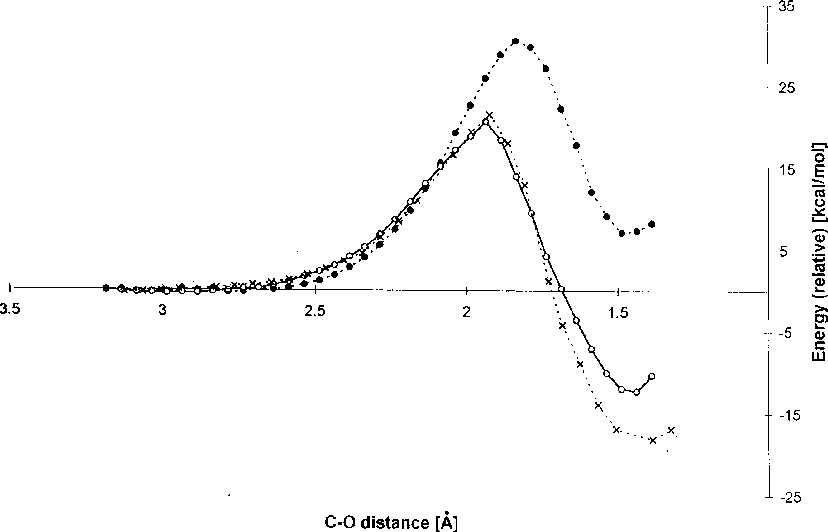 |
Reaction pathways from the MOPAC/DRIVER calculation obtained with different complexities of the input structures. A structure (Fig. 2a), closed circles and broken line; B structure (Fig. 2b) open circles and full line; C structure (Fig. 2c), crosses and broken line. The C-O distance refers to the distance of the Asp124 nucleophilic oxygen to the DCE electrophilic carbon atom. The energy (heat of formation) of the optimised geometry in each step has been calculated by the semi-empirical AM1 method.
| enzyme /input structure | experimenta | calculationb | |
| Vmax mmol/min/mg | Ea kcal/mol | delta H kcal/mol | |
| Wild type | 5,3 | 21 | -12 |
| Trp125 -> Gln | 0,15 | 26 | -4 |
| Trp125 -> Phe | 1,8 | 27 | -5 |
| Trp125 -> Arg | -c | -d | -d |
| Trp175 -> Gln | - | 31 | -4 |
| Trp175 -> Phe | -f | 28 | -4 |
Dehalogenation of the substrates 1,2-dichloroethane by wild-type haloalkane dehalogenase and tryptophan mutants: experiment versus quantum chemical calculation. (a) Michaelis-Menten kinetic constants by Kennes et al. [5] ; (b) AM1 energies from the MOPAC/DRIVER calculations; (c) no activity detectable at 10mM substrate; (d) reaction didn't proceed under used conditions; (f) not determined
An endothermic reaction was observed when only nucleophile and substrate (structure A) were included in the calculation (Fig 3). Significant changes in both thermodynamic and also kinetic characteristics of the reaction were observed when both tryptophans together with nucleophile and DCE were considered in the calculation (structure B). No significant changes in the activation energy were observed when comparing the results obtained with B and C structures, whilst a more favourable was obtained for the C structure, indicating that the rest of the active-site residues contribute to the stabilisation of the intermediate rather than the transition state.
The lower activation energy barriers were obtained for the reaction 'catalysed' by the wild type enzyme residues compared to its single point mutants (Table 1). This result is consistent with that obtained by Kennes et al. [5], who observed a very low dehalogenation activity of the same mutants in experiments with 1,2-dichloroethane. Agreement with the experiment is qualitative only, since the experimentally observed differences in the catalytic activities within mutants couldn't be reproduced.
We would like to thank Biosym Inc. for providing us the academic licence for the InsightII, Biopolymer and Discover software. We thank Academic supercomputer centers in Brno and Prague for the access to computer facilities. |