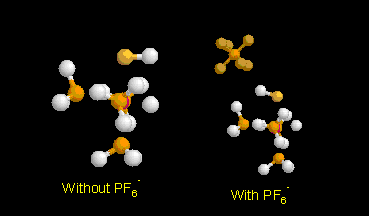
The geometry of IrH(PH3)3(SH)+ complex was optimised with and without the PF6- counter-ion. The results are dramatically different. For the isolated complex the result is similar to the IrH(PH3)3(OH)+ analogue, with the H (O-H) bending towards the hydride (Ir-H). With the introduction of the counter-ion the experimental geometry is roughly reproduced. In Figure 3 both optimised geometries are shown. Table 5 compares optimised and experimental structures for [IrH(PH3)3(SH)+] [PF6-].
Figure 3: Comparison of the molecular structures of optimized
IrH(PH3)3(SH)+ and
[IrH(PH3)3(SH)+][PF6-].
| Optimized (Å) | Experimental (Å) | Ptimized (°) | Real (°) | ||
| Ir - H2 | 1.651 | 1.642 | H12 - S - Ir | 104.6 | 110.6 |
| Ir - S | 2.467 | 2.437 | S - Ir - H2 | 79.7 | 76.5 |
| Ir - P3 | 2.378 | 2.299 | H - Ir - P3 | 77.6 | 82.7 |
| Ir - P7 | 2.509 | 2.378 | P3 - Ir - P7 | 89.8 | 82.7 |
| Ir - P13 | 2.381 | 2.354 | P13 - Ir - P17 | 178.9 | 164.3 |
| Ir - P17 | 2.381 | 2.343 | - | - | - |
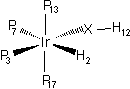
The influence of the counter-ion is clearly shown from these calculations.
The results of the Mulliken charge analysis for the optimized structure are shown in Table 6.
Ir 0.0437
H2 -0.0235
S -0.3595
H12 0.2353
F -0.8140
These results reinforce the knowledge of the influence of the counter-ion in determining the molecular structure of [IrH(PH3)3(SH)+]. It is basically an electrostatic interaction that distorts the S-H group towards the PF6- anion.
 Back to Main Page.
Back to Main Page.