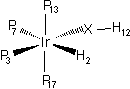
We started the study of the IrH(PH3)3(OH)+ complex by performing a geometry optimisation on the isolated cation. The results are shown on the first column of Table 2. In particular, the angle H-O-Ir stays far away from the observed one, 108.6 and 91.1° respectively.
In a second attempt at reproducing the experimental structure, we performed a geometry optimisation on the pair [IrH(PH3)3(OH)+][PF6-]. The results are shown on the second column of Table 2. The third column lists the correspondent experimental values.
| A (Å) | B (Å) | C (Å) | A (°) | B (°) | B (°) | |
| Ir - H2 | 1.647 | 1.662 | 1.711 | H12 - O - Ir | 108.6 | 106.4 | 91.1 |
| Ir - O | 2.101 | 2.119 | 2.119 | O - Ir - H2 | 93.9 | 91.0 | 92.6 |
| Ir - P3 | 2.396 | 2.374 | 2.257 | H - Ir - P3 | 84.5 | 82.5 | 77.6 |
| Ir - P7 | 2.513 | 2.532 | 2.370 | P3 - Ir - P7 | 102.0 | 107.3 | 100.1 |
| Ir - P13 | 2.413 | 2.395 | 2.337 | P13 - Ir - P17 | 177.0 | 176.0 | 158.7 |
Albeit the introduction of the PF6- anion does not improve significantly the H-O-Ir bond, the overall geometry becomes closer to the observed one.
To investigate further the nature of the interaction of the Ir-H····H-O groups, we performed a Mulliken population and density study.
The results of the Mulliken population and charge analysis are shown in Table 3.
A
B
Ir 0.1951
0.6703
H2 0.0312
-0.0226
O -0.7352
-0.7479
H12 0.3370
0.3182
OP(H····H) 0.0029
0.0146
Examination of the electronic distribution shows that the geometry of O-H and Ir-H groups are adequate for an electrostatic interaction.
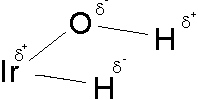
This interaction is very similar to the ones observed in typical hydrogen bonds, except for the directionality.
The overlap population between Hδ+ and Hδ- have positive, although small, values. Due to the uncertainties of these partition schemes the results should be looked at with care. Despite that fact, there is an increase in overlap population when the angle H-O-Ir diminishes.
Bond energies between IrH(PH3)3(OH)+ and PF6 were decomposed in several terms. All have been explained in detail in several occasions. In Table 4 we present such a decomposition for the formation of [IrH(PH3)3(OH)+][PF6-].
A
δEPauli -0.0063
δEElect -2.2889
δESteric -2.2952
δE(A') -0.1677
δE(A") -0.0399
δEoi -0.2076
δETotal -2.5028
As expected there is a stabilising interaction between both fragments,
the electrostatic contribution being the most significant one.
In Figure 2 the total energy, calculated with the PW91 Non-Local corrections
to the LSDA, is plotted for several H-O-Ir angles. The energy for the experimental
geometry is only 0.113 eV higher than the minimum.
Figure 2: Energy changes as function of the H-O-Ir angle.
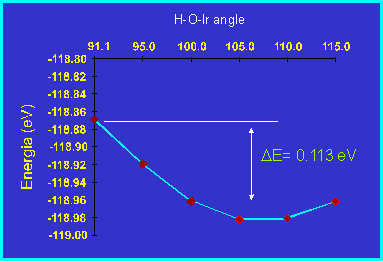
As a consequence, we conclude that all weak interactions are important
in determining the geometry of the IrH(PH3)3(OH)+ species.
 Back to Main Page.
Back to Main Page.