b Chemistry Department, Philadelphia College of Textiles and
Science,
Philadelphia, PA 19144, U. S. A.
c Lab. de Chimie Physique Moléculaire,
Faculté des
Sciences, CP 160/09, Université Libre de Bruxelles,
av. F.-D.
Roosevelt 50, B-1050 Brussels, Belgium
d Lab. of Molecular Spectroscopy, Chair of Physical
Chemistry,
Department of Chemistry, Moscow State University,
Moscow
119899, Russian Federation, C. I. S.
The general forms of the Hamiltonians for both semirigid and nonrigid models of internal rotation are essentially the same:
where j is the internal rotation angle and Veff is an effective potential containing some pseudopotential terms, which are responsible for interaction between internal and overall molecular rotations. The function F in the kinetic energy operator can be computed from the four-dimensional G matrix, elements of which are determined by atomic coordinates and masses. In the case of the semirigid model dependence F on j is due to the change in the position of the molecular center of mass during rotation of the top with respect to the frame. For the nonrigid model, along with the change of position of the center of mass, there is a parametric dependence on j for all the internal coordinates (internuclear distances, valence and dihedral bond angles). Therefore the distinction between the two models manifests itself in the character of the dependence of F on the internal rotation angle j.
The principal problem in using the nonrigid model is obtaining data on the changes in the geometrical parameters during rotation of the molecular moieties. There is no way of obtaining such information from experiment. At best, the experimental data may provide the geometry of the stable conformers corresponding to the minima on the potential curve. However, nonrigid models can be constructed on the basis of ab initio quantum mechanical optimized molecular geometries. For example, Fig. 1 illustrates the relaxation of geometrical parameters during internal rotation in the acryloyl fluoride molecule, as calculated at the MP2/6-31G* level [5]. The corresponding functions F(j) for semirigid and nonrigid models are shown on Fig. 2.
Besides the molecular geometry, ab initio calculation provides the theoretical potential function of internal rotation. As a rule, this potential function is not sufficiently accurate to describe quantitatively the torsional spectrum. However, it offer a physically meaningful initial approximation for subsequent refinement by solution of the inverse spectral problem using the set of experimentally measured frequencies. This approach has been used successfully to construct potential functions for internal rotations of isoprene [6], 1,3-butadiene [7, 8], acrolein [9], glyoxal [10], acryloyl fluoride [5, 11], 2-fluoropropenoyl fluoride [12] and nitrobenzene [13]. In the case of the acryloyl fluoride molecule the difference between nonrigid and semirigid models, which is shown on Fig. 2, is the most marked one. Although the degree of "nonrigidity", i.e. the amplitude of changes in the geometrical parameters during internal rotation, is rather significant for such molecules as 1,3-butadiene and 2- fluoropropenoyl fluoride, the semi- and nonrigid models give nearly identical results. In the case of 1,3-butadiene this is connected with the phenomenon of mutual compensation of the individual effects caused by changing each of the geometrical parameters, whereas in the 2-fluoropropenoyl fluoride molecule the top and the frame are nearly balanced. Balance decreases the effect of changes in the geometrical parameters on the F function. As for nitrobenzene, it is possible to say that the moieties of this molecule are nearly rigid. Though for some molecules a semirigid model is a reasonable approximation, this can be determined reliably only after comparison of both models. From this point of view, it is always desirable to use the the nonrigid model as a more general one. However, it is always necessary to keep in mind that the results obtained from the nonrigid model are also significantly dependent on the level of the quantum mechanical calculation providing the character of the molecular geometry relaxation. See Ref. 8 for a more detailed consideration of practical use of the nonrigid model.
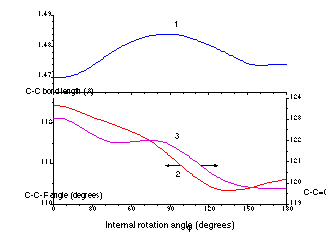
Fig. 1. Ab initio geometrical parameters of the acryloyl fluoride, CH2=CH--CF=O, as functions of the internal rotation angle j.
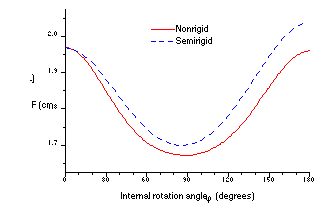
Fig. 2. Comparison of the functions F(j) for semi- and nonrigid models of internal rotation in acryloyl fluoride as calculated using quantum mechanical data obtained at the MP2/6-31G* level.
2. R. Meyer, E. B. Wilson, J. Chem. Phys., 53 (1970) 3969.
3. R. Meyer, J. Mol. Spectrosc., 76 (1979) 266.
4. M. A. Harthcock, J. Laane, J. Mol. Spectrosc., 91 (1982) 300.
5. G. R. De Maré, Yu. N. Panchenko, A. V. Abramenkov and Ch. W. Bock, Can. J. Chem., 71 (1993) 656.
6. Yu. N. Panchenko, V. I. Pupyshev, A. V. Abramenkov, M. Traetteberg and
S. J. Cyvin, J. Mol. S
Return to main WATOC Poster page