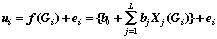 ,
i = 1, ..., N; (1)
,
i = 1, ..., N; (1)
INTRODUCTION. Construction of quantitative "structure-property" relationship (QSPR) within the framework of different mathematical approaches and methods [1-3] has the aim to determinate the chemical structures, which is "active for the given QSPR-model". It means, that the QSPR-model, receiving such structure, will produce a desired high value of its activity [4,5]. If the QSPR-model has many parameters and functional relationship is non-linear or combines several function (models), then the practical solution of the "inverse task" can be achieved by the "QSPR computer screening" of chemical compounds selected from classes, which are interesting for the chemist. The complexities of the molecular generation task consist in that, what the sequence of compounds for screening should satisfy two contradictory conditions. On the one hand, limited computing resources force to decrease the number of structures, which put to the QSPR-model. At the same time, it is desirable to receive structures of new chemical classes. Thus, it is actual task to develop the new methods of molecular generation, which produce "potentially active" structures, which can possess the desired property with high probability.
The known molecular generation methods used the combinatorial exhausting enumeration (with given restriction) of all structures that have the specific brutto-formulas [6,7]. Other method assumes active involvement of the user, which must define the molecular "core" - a base fragment of all generated molecule[8,9]. Then he puts the substitute's lists and indicates the core's atoms to which the substitute must be assign. The method has the essential shortage because the class of obtained compounds strictly determine by the chosen "core" fragment.
In this work we describe a molecular generation method for a solution inverse QSPR task that develops the approach, offered in [10,11]. The approach uses the evolution algorithm for the construction a minimum length graph's sequence, which connect "start" and "target" molecular graphs. These graphs are chosen from the learning data base (DB) and must belong to the "active" structures.
STATEMENT OF THE TASK. The molecular graph (M-graph) is simple graph
G= (V (G), E (G)) without loops, which have the following specific
features[1,2,12,13]:
1. The graph's vertices (set V(G)) have symbol labels, which are the names of the elements in the periodic table.
2. The graph's edges (set E(G)) have symbol labels, which denote the type of (covalent) chemical bond: ordinary (single) bonds, double bonds, triple bonds or aromatic bonds.
3. The hydrogen atoms are not vertices of graph (M-graph "with erased hydrogen").
4. The degree of vertices does not exceed six.
5. The graph's vertices have additional attributes: (1) number of hydrogen atoms joined to vertex; (2) the charge of atom; (3) the space localization of vertex - (X, Y, X) coordinates.
Two M-graphs G1 and G2 are said to be isomorphic (G1 [[congruent]] G2), if there is exists a one-to-one correspondence of their vertices such, that the correspondence of edges is saving, and the labels of appropriate vertices and edges coincide. The coincidence of attributes of vertices is optional and required only in especially stipulated cases.
Suppose a learning data base (DB) containing N records (entries) downloaded to the computer. Every i-th entry contains a pair (Gi, ui), where Gi is a M-graph, represented by an atomic connectivity table (completed with all the necessary atom and bond labels); ui is the numerical value of an experimental property of the substance whose molecules are represented by the M-graph Gi. Let a model of a QSPR - property estimation constructed as linear function f of the M-graph features, which minimized a squared error sum[2,3,12,13]:
,
i = 1, ..., N; (1)
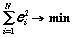 ;
(2)
;
(2)
where N is DB size; ei is the property estimation error for the i-th graph, Xj (G) is the value of the j-th descriptor of the graph G; bj are the coefficients found from the condition (2).
For the graph metric definition we must to represent each M-graph G by a features vector. Let the set of the M-graphs fragments H = { hj }, ( j = 1,..., L) is given. A structural descriptor Xj(G) is defined as a number of the fragment hj repetition in the graph G. According to the M-graphs isomorphism definition we consider that the fragment hj enters in graph G, if there is exist such subgraph G' Í G, that hj [[congruent]] G'. A vector (X1,X2,..., XL) is said to be a structural spectrum of graph G relatively H. A structural spectrum allows to define one of the following graph metrics (in the first approximation all weight factors wj =1):
1. Rectangular (Manhattan) Metric: 2. Euclidean Metric:
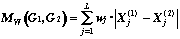
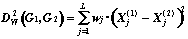
3. Hamming Metric:
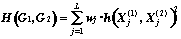 ,
and
,
and
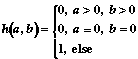
4. Metric based on structure's similarity:
à)
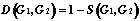
á)
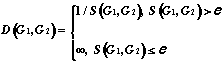 and
and
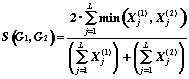
Let two "active" structures: GS (the start graph) and GT (the target graph) are selected from learning DB. With the help of evolution algorithm [15] we shall construct the list of structures Gi, such that:
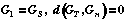 ;
;
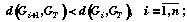 (3)
(3)
The sequence { Gi } is an outcome of the algorithm
GENERATION ALGORITHM. The initial parent-graph set LP1 contain only one start graph GS. Let define on the molecular graphs the following elementary operations { Ti }: to add or to delete an edge, to add or to delete atom, to rename atom, to change a type of bond. We shall describe these operations in the supposition, that V(G), E(G) are the vertices and edge's sets of a graph G, and p(v) is the degree of vertex v, GT is a target graph..
TDE: : Edge deleting operation: G2=TDE(G1);
V(G2) = V(G1); E(G2) = E(G1) \ { e }, e [[propersubset]]E(G1)
Restriction: the graph connectivity should not be broken, i.e. only ring edges can be deleted;
Probability of application PDE :
PDE = 0, if | E(GT) | > | E(G1) |;
PDE = ( | E(G1) | _ | E(GT) | + 1 ) / | E(GT) |, if | E(GT) | < | E(G1) |,
where | E (G) | denotes the number of edges in the graph G.
TDV1: A "degree 1" vertex deleting operation with its edge: G2=TDV1(G1);
V(G2) = V(G1) \ { v }, v [[propersubset]]V(G1); p(v)=1
E(G2) = E(G1) \ { (v,u) }, (v,u) [[propersubset]]E(G1), u [[propersubset]]V(G1);
Probability of application PDV1: PDV1 =0, if | V(GT) | > | V(G1) |;
PDV1 = ( |V(G1) | _ | V(GT) + 1 | ) / | V(GT) |, if | V(GT) | < | V(G1) |,
where | V(G) | denotes the number of vertices in the graph G
TDV2: A "degree 2" vertex deleting operation with one its edge: G2=TDV1(G1);
V(G2) = V(G1) \ { v }, p(v)=2;
E(G2) = ( E(G1) + { (u,w) } ) \ { (v,u), (v,w) },
where v, u ,w [[propersubset]]V(G1); (v,u), (v,w) [[propersubset]]E(G1),. (u,w) [[reflexsubset]]E(G1)
Restriction: the edges (v,u), (v,w) have ordinary bond type and p(v)=2.
Probability of application PDV2 : PDV2 = PDV1 .
TIE: An edge insertion operation gor the vertex v: G2=TIE(G1);
V(G2) = V(G1); E(G2) = E(G1) + { (v,u) },
where v, u [[propersubset]]V(G1), (v,u) [[reflexsubset]]E(G1).
Restriction: in graph G2 p(v), p(u) should not exceed valences of v, u atoms
Probability of application PIE : PIE=0, if | E(GT) | < | E(G1) |;
PIE = ( | E(GT) | _ | E(G1) | + 1 ) / | E(GT) |, if | E(GT) | > | E(G1) |,
TIV1: A "degree 1" vertex insertion operation : G2=TIV1(G1);
V(G2) = V(G1) + { v }, v [[reflexsubset]]V(G1); p(v)=1;
E(G2) = E(G1) + { (v,u) }, u [[propersubset]]V(G1).
Probability of application PIV1 : PIV1 =0, if | V(GT) | < | V(G1) |;
PIV1 = ( |V(GT) | _ | V(G1) | + 1 ) / | V(GT) |, if | V(GT) | > | V(G1) |,
TIV 2: A "degree 2" vertex insertion operation : G2=TIV2(G1);
V(G2) = V(G1) + { v }, p(v)=2;
E(G2) = ( E(G1) \ { (u,w) } ) + { (v,u), (v,w) },
where u, w [[propersubset]]V(G1); v [[reflexsubset]]V(G1); (v,u), (v,w) [[reflexsubset]]E(G1); (u,w) [[propersubset]]E(G1).
Probability of application PIV2 : PIV2 = PIV1
TME: Edge moving operation: G2=TME(G1);
V(G2) = V(G1); E(G2) = ( E(G1) \ { (v,u) } ) + { (v,w) };
where (v,u), (u,w) [[propersubset]]E(G1); (v,w)
[[reflexsubset]]E(G1),
Restriction: p(u) > 3.
Probability of application PME : PME =0, if (| E(GT) | != | E(G1) |) or (p(u) = 2);
PME = ( p(u) -2 ) / | E(GT) |, if (| E(GT) | = | E(G1) |) and (p(u) > 2).
TLV: Vertex relabeling operation : G2=TLV(G1);
V(G2) = V(G1), E(G2) = E(G1), VLabel( v,G1 )=A1, VLabel( v,G2 )=A2, A1 != A2;
where VLabel(v,G) is the label of the vertex v in graph G;
Probability of application PLV :
PLV = 0, if #VLabels( A1,G1 ) = #VLabels( A1,GT )
PLV = | #VLabels( A1,G1 ) - #VLabels( A1,GT ) | / #VLabels( A1,GT ),
if #VLabels( A1,G1 ) != #VLabels( A1,GT );
where #VLabels( A1,G ) is the number of the A1 labeled vertices in the graph G.
TLE: Edge relabeling operation: G2=TLE(G1);
V(G2) = V(G1), E(G2) = E(G1),
ELabel( e,G1 )=E1, ELabel( e,G2 )=E2, E1 != E2;
where ELabel( e,G ) is the label of the edge e in the graph G;
Probability of application PLE :
PLE = 0, if #ELabels( E1,G1 ) = #ELabels( E1,GT )
PLE = | #ELabels( E1,G1 ) - #ELabels( E1,GT ) | / #ELabels( E1,GT ),
if #ELabels( E1,G1 ) != #ELabels( E1,GT );
where #ELabels(E1,G) - is the number of the E1 labeled edges in the graph G.
The transformation of the parent-graph Gj is the following.
To each vertex v of graph Gj the following operation
with appropriate probabilities will be applied: insertion vertex TIV1,
TIV2; deleting vertex TDV1, TDV2, insertion of an edge TIE
and relabeling of vertex TLV. To each edge e of graph
Gj the following operation with appropriate probabilities are
applied: insertion an edge TIE, deleting an edge TDE:, moving of
an edge TME and relabeling of an edge TLE. If the application of
any operation has taken place, we receive the child-graph
G'j+1. If a condition
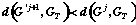 is true the we add child-graph to the list of offspring
LGj+1, and we continue examination / transformation of
vertices and edges of the parent-graph. We stop building of the list
LGj+1 after encounting all vertices and edges of
parent-graph Gj
is true the we add child-graph to the list of offspring
LGj+1, and we continue examination / transformation of
vertices and edges of the parent-graph. We stop building of the list
LGj+1 after encounting all vertices and edges of
parent-graph Gj
Let on j-th step the parents-graph list LPj = {
Gjk k=1,..., Q} is generated. The developing
of k-th parent-graph forms a generation branch. The
transformation of graph Gjk generates the list of
offspring-graphs LGj+1k . After handling all branches
we build the common list of all offspring-graph of the (j+1)-th
step:
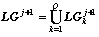 .
.
If Q is a size of a population, then we select from
LGj+1 list only the Q "best" graphs in a
sense of similarity to target graph GT and build the parent-graphs list
of the next step LPj+1. The condition (3) guarantees
the "convergence" of the graph sequence to target graph GT. A
construction of the graph Gn : d(Gn,GT) = 0 is a "stop condition" of
evolution algorithm. An outcome of generation will be the set of all the
parents-graphs LPj:
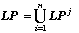
For an effective algorithm realization one can introduce a series of restrictions, directed to avoid of certainly unacceptable graph transformations: (1) to not suppose modification of graph, which violate the atom's valence; (2) at the edges deleting the graph connectivity must be preserved.
FORMING THE FRAGMENTS SET H. One of the approaches to a solution of this task is the full fragments enumeration in all M-graphs of learning DB. As enumerated fragment type we use linear atom's chains of length q (q-chains, q=1,2,3,...). A result of such enumeration is a pair (H, X), where H = {hj, j=1,..., L} is a set of all nonequivalent q-chains, found in learning DB, X= || Xij ||, (i=1,..., N, j=1,..., L) is a "molecule-chain" matrix, where Xij is the number of recurrings a j-th q-chain in the i-th molecule; N - size of the learning DB; L - number of found fragments (as a rule, N<<L). The i-th row of this matrix represents a structural spectrum of graph Gi.
For the fine fragments distinguishing atom's markers will be introduced, aimed tp classify atom's topological and chemical singularities [17]. An atom degree(i.e. number of incident edges) will be encoded as p-marker, which can accept (for the connected graphs of organic structures) six values { 1, 2, 3, 4, 5, 6 }. From Figure 1 it is visible, that one p-marker it is not enough for distinguishing "valence environment" of the carbon atom. The b-marker will encode types of atom's chemical bond as follows:
"s" (single) - when the atom has only single bonds;
"d" (double) - when the atom has a double bond;
"t" (triple) - when the atom has a triple bond;
"w" - when the atom has two double bonds;
"a" (aromatic) - when the atom has an aromatic bond.
An extended atom's labels (with the b- and r-markers) are the following strings: "NNpb". On the Figure 2 the extended atom's labels are present with the codes of diatomic chains (1-chains) ,which are formed by the various "carbon - carbon" bonds.
CH3 - - CH- CH2= > CH - - CH =
p=1 b="s" p=2 b="s" p=1 b="d" p=3 b="s" p=3 b="d"
CH[[equivalenc > C < > C = = C = -
e]] C[[equivalence
]]
p=1 b="t" p=4 b="s" p=3 b="d" p=2 b="w" p=2 b="t"
Fig.
1. Distribution of the carbon atom's valency and the corresponding values of
atom's markers: p-marker (a degree of atom); b-marker (a chemical
bond "s" - all bonds are single; "d" - there is the double bond; "t" - there
is the triple bond; "w" - there are two double bonds).

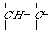
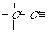 "C_4sC_4s" "C_3dC_4s" "C_2tC_4s"
"C_4sC_4s" "C_3dC_4s" "C_2tC_4s"
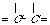
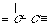
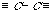 "C_3dC_3d" "C_2tC_3d" "C_2tC_2t"
"C_3dC_3d" "C_2tC_3d" "C_2tC_2t"
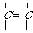
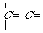 =C=C=
"C_3dC_3d" "C_2wC_3d" "C_2wC_2w"
=C=C=
"C_3dC_3d" "C_2wC_3d" "C_2wC_2w"
Fig.
2. Various "carbon-carbon" bonds (1-chain) and their codes as a string "A1A2",
where A1, A2 are extended atom's label (without a r-marker), "Ñ_"
- the name of carbon atomAn edge is said to be a ring edge, if at its deleting the graph connectivity of is not breaked, and chain (acyclic) - otherwise. If the atom is incident to the ring edge, we said it is a ring atom. Among "ring" atoms we distinguish "purely ring" atoms, (i.e. atoms, which have only ring edges), and "ring with the substitute" atoms, which have an acyclic edge. One can use a r-marker (for an extended label formation), which have the following definition:
"c" (chain) - an atom is an acyclic atom;
"r" (ring) - an atom is a "purely ring"; atom;
"s" (substitute) - an atom is a "ring with the substitute" atom.
So, the extended labels look like: "NNpbr", where NN is an atom's name. Two q-chains is said equivalent, if they have the identical chain's code. Chain's code is a sequence of extended labels of atoms, which form a q-chain. From two possible chain codes a more lexicographic higher strind is selected. Thus, for the distances calculation between graphs the system carry out extended labelling of atoms in the parents-graphs and in the offsprings-graphs. This allows to take into account fine differences between structural spectrum fragments (q-chains).
METRIC ADAPTATION. The evaluation of weight coefficients wj can also be carried out with help of evolutionary algorithm [16]. For this purpose it is necessary to define a function of metric estimation and to determine the coefficient's variation rules from step to step. Let to estimate the metric quality with the help of the cluster analysis of M-graphs distributed in the given metric spase (i.e. after decomposition of the DB's graphs into non-intersecting groups):
Let the learning DB is divided into P clusters. The Shannon
information function as the estimate criterion is defined as follow. Suppose,
there are N+p and N-p objects of
first ("active") and second ("inactive") class in p-th cluster,
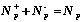 .
.
Then the estimate value for the p-th cluster is
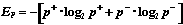 ,
,
where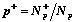 ,
,
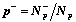 ( Ep = 0, if p+ = 0 or
p-- = 0 ).
( Ep = 0, if p+ = 0 or
p-- = 0 ).
Let define the global metric estimate function E as:
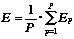 .
.
We can see, that function E achieves its maximum value, equal to unit, if and only if exactly one half of objects in each cluster is from first class and one half - from the second class (p+ = p-- = 0.5). Minimum value of function E, equal to zero, is reached, if all molecules in each cluster are from the same class (we say clusterization has appeared completely successful).
This function E had to be minimized by selecting the weight
coefficients wj. Let vector
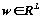 is "gen", which define a solution of the metric selection problem. Mutation
operation changes one of its coefficients according to the formula:
wj(q + 1) = wj(q) + Dw,
where q is the step count, and
0 < Dw < 1. Crossover operation is
standard[15].
is "gen", which define a solution of the metric selection problem. Mutation
operation changes one of its coefficients according to the formula:
wj(q + 1) = wj(q) + Dw,
where q is the step count, and
0 < Dw < 1. Crossover operation is
standard[15].
The practical realization of proposed graph generation algorithm needs the fast distance calculation between the current and terget graphs. Thus the most critical parameter becomes L - the number of fragments, participating in the metric definition. This number can be reduced by the preliminary analysis of the "molecule-fragment" matrix. First, it is needed to delete all its columns-constant. Secondly, we can consider high correlated columns as one generalized column, because their contribution to the metric will be about identical. Such columns could be deleted from the matrix, and their average vector column is added. Some phases of the considered method (decomposition of M-graphs into line atoms' chains (fragments), "molecule-fragment" matrix construction, selection of mutually correlated column) are realized in the complex QSPR system BIBIGON [12,13,17].
As show above the evolution algorithm was applied twice: in the beginning the genetic algorithm can be used for the metric optimization (if needed), which became adequate for M-graph's property classification (as "active" or "inactive" structures), and then evolution algorithm used to transform and select the graphs, which form the of minimal length path in this metric.
The offered approach to the molecular graph's generation will be tested for the possibility to build "active" compound of different chemical classes. Thus, the preservation of structural similarity of the generated graphs with given "start" and "target" ones must provide the probability that the inverse task will be successfully solved.
This study was supported by the Russian Foundation for Basic Research (project nos. 96-01-01598 and 94-01-00041)
References
1. Chemical application of topology and graph theory. / Ed. by R.B.King, - Studies in Physical and Theoretical Chemistry, vol. 28, Elsevier, Amsterdam, 1983, 560 p..
2. Kier L.B., Hall L.H. Molecular connectivity in structure-activity analysis. Willey: London, 1986, 251p.
3. Stuper A.J., Brugger W.E., Jurs P.C. Computer Assisted Studies of Chemical Structure and Biological Function / New York: Wiley, 1979.
4. Skvortchova M.I., Baskin I.I., Slovohotova O.A., Palyulin V.A., Zefirov N.S. The inverse task in the QSAR/QSRP-analysis for the topological indexes, describing the molecular form (an Kier index) / Bull. of Russian Acad. Sci., 1992, v.324, n.2, p.344-348.
5. Baskin I.I., Gordeeva E.V., Devdariani R.O., Zefirov N.S., Palyulin V.A., Stankevich M.I. The methodology of the inverse task solution in a problem a structure - property for some topological indexes. / Bull. Acad. Sci. USSR, 1989, v. 307, p.613--617.
6. Molodtchov S.G., Piotuh-Peletchkii V.H.. The designing of the nonisomorphic graphs by the given set of structural parameters. / In " Algorithms of the analysis of a structural information" ,- The Computing Systems, Novosibirsk, v.103, 1984, ñ. 51.
7. Kvasnicka V., Pospichal J. // J.Chem.Inform. Comp.Sci., 1990, vol.30, p.99.
8. Kumskov M.I., Sukhachev D.V., Palyulin V.A., Lomova O.A. Fast generation of molecular graphs with the help of the base structures in a Wiswesser code / Abstracts of InterUniversity Conf. "Molecular graphs in chemical researches", Kalinin, Russia, 1990, p.54-55. (in Russian)
9. Lomova O.A., Sukhachev D.V., Kumskov M.I., Palyulin V.A., Tratch S.S., Zefirov N.S. The Generation of Molecular Graphs for QSAR Studies by the Acyclic fragment combining / Commun. Mathem.Chem. (MATCH), 1992, n.27, p.153-174.
10. Johnson M., Naim M., Nicholson V., Tsai Ch.-C. Unique Mathematical Features of the Substructure Metric Approach to Quantitative Molecular Similarity Analysis / In: "Graph Theory and Topology in Chemistry", Eds. by R.B.King and D.H.Rouvray - Studies in Physical and Theoretical Chemistry, vol. 51, Elsevier., Amsterdam, 1987, p.219-225.
11. Kumskov M.I., Mityushev D.F. Solving the Inverse Problem of Molecular Graph Recognition by Constructing Minimal Paths in a Labeled Graph Space / Pattern Recognition and Image Analysis (Interperiodica Publishing, Russia), 1996, vol.6, n.2, p.277-278.
12. Kumskov M.I., Mityushev D.F., Oshchepkov E.N., Kolovanov E.A. Computer-Aided Extraction and Application of Knowledge about Structure-Property Relationship in Factographical Chemical-Structure Data Bases / Pattern Recognition and Image Analysis (Interperiodica Publishing, Russia), 1995, vol.5, n.4, p.589-601.
13. Kumskov M.I., Zyryanov I.L., Svitan'ko I.V. C A New Method for Representing Spatial Electronic Structures of Molecules in the Problem of Structure _ Biological Activity Relationship / Pattern Recognition and Image Analysis (Interperiodica Publishing, Russia), 1995, vol.5, n.3, p.477-484.
14. Kohov V.A.. Method for the Quantitative Similarity Graph Definition on the Base of the Structural Spectrum / Bull. Russian Acad. Sci., "Technical Cybern" Series, 1994, n.5, p.143-159. (in Russian)
15. Davis L. Handbook of genetic algorithms / Van Nonstrand Reinhold, New York, 1991.
16. Yurchenko Ò.À., Kumskov M.I., Vorona D.G. Metric Selection for Properties of Chemical Compounds Classification with the Help of Genetic Algorithm. / Extended Abstracts of the 4-th Open German-Russian Workshop "Pattern Recognition and Image Understanding", Valday, Russian Federation, 1996.
17. Kumskov M.I., Ponomareva L.A., Smolenskii E.A., Mityushev D.F., Zefirov N.S. Automatic Formation Method for Structural Descriptors of Organic Compounds for Quantitative Structure-Property Relationships./ Russian Chem. Bull., 1994, vol.43, n.8, p.1317-1319.