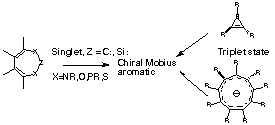
The presence of a Möbius configuration of pp atomic orbitals is associated with the presence of a chiral symmetry element (e.g. C2 axis) and the absence of a Cs plane of symmetry in those molecules which could have either (or both). Our criteria for judging aromaticity in such species are two fold. Nucleus independent chemical shifts5 approximately associate overall negative NICS values with a diatropic ring current and aromaticity, and positive values with a paratropic ring current and anti-aromaticity, although the integrated NICS value contains contributions from both types of ring current. We emphasise here not the absolute NICS values, but their trends. The second criterion derives from the original suggestion by Heilbronner that an ideal Möbius aromatic would exhibit neither bond length localisation, nor any localisation of the Möbius twist present in the ring. We have previously noted6,7 that ring perfluorination enhances such delocalisation in other Möbius systems and reduces the mixing of C-R s components with the cyclic pp array, and have also included examples of F-substituted systems. Optimised energies at the RB3LYP//6-31G(d) level8 of a variety of ring systems derived from 1 are listed in Table 1, and of triplet state monocyclic ring systems at the ROB3LYP//6-31G(d) level in the Table 2.¶
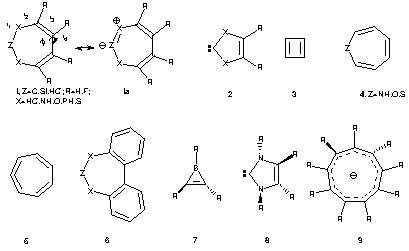
The diaza system 1, X=NH, R=H, Z=C: is the 4n homologue of the carbene 2, X=NMe, a ligand of current interest as a phosphine equivalent in metal coordination chemistry.9 Initial calculations at a planar geometry reveal it to be strongly anti-aromatic, with a NICS value quite similar to that calculated for cyclobutadiene 3. The two C-N bond lengths (1.35Å) are equal and their value indicates that a closed shell ylid representation (i.e. 1a) is better than the carbene. Unlike 3 however, no first order Jahn-Teller distortion to the bond-alternating form represented by 1a occurs, because the presence of the two nitrogen heteroatoms increases the energy separation of the otherwise degenerate orbitals. Given the presence of two nitrogen lone pairs, we were quite surprised to find that only one, small negative root was calculated for the Hessian matrix at the planar geometry (Table 1), the vectors of which do not correspond to pyramidalisation of the lone pairs, but instead represent a twist to create a chiral species with C2 symmetry. The energy lowering resulting from following this small geometric distortion is an inconsequential 0.15 kcal/mol, but this is accompanied by a relatively large reduction in the calculated NICS value of 4.2 ppm. The angular ring strain is the factor that prevents more Möbius distortion, and hence it appears that these systems are finely balanced between planar Hückel anti-aromaticity and Möbius aromaticity; in this specific case the former still imparting the dominant character. We also contrast this distortion with that found experimentally and theoretically for the 8-p azepine, oxepine and thiepine rings (4), for which a Cs distorsion is of lower energy than a C2 mode.10 A closer analogy can be drawn to the 8-p system 5.7 Another stabilising mode exhibited by anti-aromatic systems is aromatisation by p-p* excitation to a lower energy triplet state, as occurs for e.g. 3. This mode is not adopted by 1 (Table 1), the triplet p-p* state being significantly higher in energy than the singlet, in agreement with its ylid (1a) rather than carbene-like characteristics. The aromatic NICS value of the triplet is appropriate for a 4n pp triplet.11 The perfluoro system 1, X=NF, R=F, Z=C:, which is significantly more twisted, as indicated by the dihedral angle f, and is less anti-aromatic, as measured by the NICS value and reduced bond alternation. The chiral system derived from biphenyl (6) is likewise only mildly anti-aromatic, and a viable candidate for synthesis.
The dioxa system 1, X=O, R=H, Z=C: as a planar ring also reveals a single imaginary vibrational mode distorting to C2 symmetry. In this case the reduction in energy is rather greater (1.5 kcal/mol), the ring twists more, and the NICS value and bond alternation both reduce more than the diaza system (Table 1). Perfluorination also induces a negative NICS value corresponding to some aromatic character and the bond alternation reduces even further. Disproportionation to CO2 and two units of alkyne is only modestly exothermic [-12.4 kcal/mol at B3LYP/6-311G(3d) level], and perfluorination changes this to an endothermic value [+52.9 kcal/mol at B3LYP/6-31G(d)]. The latter in particular therefore is most unlikely to easily fragment, and may well therefore be quite stable thermodynamically. The corresponding values for X=NH/NF were an even more emphatic +62.2 and +124.1 kcal/mol respectively.
Of the various other X, R and Z substituents investigated¶ the following are noteworthy. The combination Z=CH+, X=CH- corresponds to C7H7-, which is reported here in Möbius form. First-order Jahn-Teller induced bond alternation (i.e. as with 1a) breaking the C2 symmetry can be seen, a feature not present with the more Möbius-aromatic perfluoro form. The lowest energy form of 1, X=PH, R=H, Z=C: has a Möbius distortion but another higher energy isomer with "tub" like Cs symmetry can also be located. In contrast, 1, X=S, R=H, Z=C: reveals neither C2 nor Cs distorsion, the planar C2v form revealing neither any calculated negative roots for the Hessian nor any symmetry-breaking bond alternation. This behaviour is similar to that reported12 for completely planar cyclic 8pp (tetrakis(bicyclo[2.1.1]hexeno) cyclooctatetraene, which also sustains not only a paratropic ring current but also exhibits large bond length alternation (1.33, 1.50Å). Perfluorination of 1, X=S, R=F, Z=C: however induces Möbius distortion and a large (-16ppm) aromatisation of the NICS value. Replacing Z=C: with the silylene equivalent (Z=Si:) similarly results in C2 distortions and tends to produce less anti-aromatic species (Table 1).
The first entry (Table 2) for triplet excited state Möbius aromatics corresponds to difluoro borirene13 (7), a neutral boron substituted analogue of cyclopropenium cation. As a closed shell singlet, 7 is planar and aromatic, with C2v symmetry.13 The open shell triplet 3A state retains only the C2 axis of symmetry and the NICS value indicates aromaticity rather than anti-aromaticity, as accords a 4n+2 Möbius aromatic. We emphasize that at this stage, we are not claiming that the Möbius triplets are always the most stable triplet possible, merely that in many cases a triplet with such symmetry can be located. The optimised geometry of the all-carbon cyclopropenium 3A cation also has C2 symmetry. No NICS value is computed but some bond length alternation does occur (1.412, 1.503Å), indicating this species is not highly aromatic. Curiously, 6 pp examples for this type are sparse. A 3A triplet state of furan shows a distortion towards C2 symmetry, and the NICS value is indicative of mild aromaticity, as are 8, R=F and the phosphorus analogue. The 6-electron 3A triplet perfluoro tropylium cation optimises to a planar (C2v) non-Möbius form.
Not all the larger ring examples are conspicuously aromatic, probably for a variety of reasons. For example, the NICS probe is placed at a computed ring centroid, and the conformation of some systems ensures that the fluorine ring substituents can also approach this position, thus perturbing the NICS value. Furthermore, boron, introduced to replace C+ in some models, is also known to inhibit aromaticity.14 The most prominently aromatic triplets were identified for the 10 (9) and the 14 pp systems. Thus triplet 9 exhibits minimal bond alternation¶ 1.408, 1.407, 1.396, 1.377, 1.411Å and small dihedral angle alternation between adjacent pairs of ring atoms (25.0, 8.0, 33.0, 36.0). The finite size of the ring, and the presence of one pseudo trans ring component, precludes a total absence of alternation. Similarly, the 14 electron triplet system C12F122- manifests only two C-C bond lengths; 1.377 and 1.404Å.
We conclude that ring systems such as 1 are predicted to attenuate the Hückel anti-aromaticity expected of a planar 4n pp system with varying degrees of C2 symmetric distorsion towards Möbius aromaticity. We also propose the first formal examples of chiral Möbius conformations of 4n+2 electron triplet aromatic annulenes corresponding to the last of the four aromaticity rules derived from simple Hückel molecular orbital theory, and suggest that these may be of interest in emerging applications such as chiral photochemistry.15
| Table 1. Calculated energies
[RB3LYP/6-31G(d),a Hartree] and NICS values [B3LYP/6-31G(d), ppm] for 1 and 6. |
||||
|---|---|---|---|---|
| 1 | Energy | NICS(0)/GIAO | r1,r2,r3,r4,f | Point Group |
| Z=C:, X=NH, R=H | -303.5375a | 21.1 | 1.360,1.428,1.337,1.468, 14.3 | C2 |
| Z=C:, X=NH, R=H | -303.6241b | 23.6 | 1.352,1.425,1.331,1.467, 0.0 | C2vc |
| Z=C:, X=NH, R=H | -303.6243b | 19.4 | 1.354,1.425,1.331,1.465, 18.4 | C2 |
| Z=C:, X=NH, R=H, triplet | -303.5792b | -8.3 | 1.375,1.370,1.417,1.373, 0.0 | C2v |
| Z=C:, X=NH, R=F | -700.4496 | 1.2 | 1.367,1.410,1.366,1.450, 33.2 | C2 |
| 6, Z=C:, X=NH | -610.8548 | 8.9 | 1.360,1.426,1.409,1.487, 35.7 | C2 |
| Z=C:, X=O, R=H | -343.3315b | 15.2 | 1.333,1.407,1.325,1.456, 0.0 | C2vd |
| Z=C:, X=O, R=H | -343.3340b | 7.7 | 1.332,1.405,1.326,1.452, 24.9 | C2 |
| Z=C:, X=O R=H | -343.2339e | 8.8 | 1.345,1.405,1.333,1.456, 23.7 | C2 |
| Z=C:, X=O, R=F | -740.1387 | -1.3 | 1.353,1.379,1.334,1.448, 29.0 | C2 |
| 6, Z=C:, X=O | -650.5536 | 6.3 | 1.343,1.410,1.400,1.474, 32.2 | C2 |
| Z=CH+, X=HC-, R=H | -270.8684 | 79.4 | 1.404 (1.393), 1.452 (1.465), 1.356 (1.350), 1.500, 25.4 |
C1 |
| Z=CH+, X=HC-, R=F | -965.5308 | -7.5 | 1.384, 1.421, 1.347, 1.450, 48.4 | C2 |
| Z=C:, X=PH, R=H | -876.6275 | 41.3 | 1.673,1.660,1.480,1.340, 0.0 | Cs |
| Z=C:, X=PH, R=H | -876.6920 | 7.1 | 1.707,1.830,1.348,1.469,35.7 | C2 |
| Z=C:, X=S, R=H | -989.1832 | 18.4 | 1.654,1.820,1.340,1.470, 0.0 | C2v |
| Z=C:, X=S, R=F | -1386.0821 | 2.3 | 1.654,1.826,1.338,1.459,38.4 | C2 |
| Z=Si:,X=NH, R=H | -555.0148 | 14.0 | 1.748,1.414,1.344,1.464,23.69 | C2 |
| Z=Si:, X=O, R=H | -594.7732 | 7.4 | 1.678,1.375,1.341,1.462,22.9 | C2 |
| Z=Si:, X=PH, R=H | -1128.1596 | 4.7 | 2.256,1.828,1.348,1.465, 46.9 | C2 |
| Z=Si:, X=PH, R=H | -1128.0819 | 25.5 | 2.254,1.670,1.470,1.340, 0.0 | Cs |
| Z=Si:, X=S, R=H | -1240.6793 | 5.2 | 2.178,1.790,1.330,1.460, 50.0 | C2 |
| Table 2. Calculated energies
[ROB3LYP/6-31G(d),a Hartree] and NICS
values [UB3LYP/6-31G(d), ppm] for perfluoro substituted 3A state Annulenes. |
|||
|---|---|---|---|
| Annulene ring Size | Charge/heteroatom | 3A Energy | NICS(0)/GIAO |
| 2-electron systems | |||
| 3 | B | -400.4222 | -9.7 |
| 3 | +1 | -411.4413 | - |
| 6-electron systems | |||
| 5 | O | -626.8068 | -3.7 |
| 5 | 2, X=O | -464.2103 | -0.7 |
| 5 | 2, X=S | -1110.1515 | -0.3 |
| 5 | 2, X=NF | -622.8091 | -5.0 |
| 5 | 2, X=PF | -1196.2361 | -3.6 |
| 7 | B | -952.26327 | 2.6 |
| 7 | +1 | -961.8441b | - |
| 8 | 1,2-B,B | -1076.9976 | -0.6 |
| 9 | 1,4,7-B,B,B | -1201.7703 | -1.1 |
| 10-electron systems | |||
| 8 | -2 | -1103.2830 | -3.4 |
| 9 | -1 | -1241.3459 | -13.0 |
| 10 | - | -1379.1909 | -3.4 |
| 11 | B | -1503.9514 | -1.4 |
| 12 | 1,6-B,B | -1628.7290 | -3.8 |
| 14-electron systems | |||
| 12 | -2 | 1655.0680 | -12.1 |