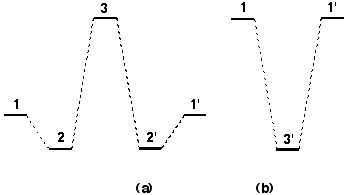
Figure 1. (a) Normal potential, (b) inverted non-classical potential
In addition to the ubiquitous proton transfer reaction, which has been extensively studied for many years,1 several other atom or group transfer reactions have recently proven to be of considerable synthetic and theoretical interest. Oxygen atom or NR group transfers from species such as oxirane, dioxirane or oxaziridine to an alkene are synthetically important, and can often exhibit complex stereoelectronic and stereoselective behaviour.2 The potential energy profiles for such atom transfer reactions can fall into two basic types (Figure 1).
Figure 1. (a) Normal potential, (b) inverted non-classical potential
The normal type (a), exhibited by most proton transfers between an acid and a base, and oxygen atom transfers such as between an alkene and an oxirane, involves the formation of a complex 2 or 2' between the atom or proton donor and acceptor. An energy maximum 3 occurs when the atom is equidistant between the two exchanging species. The angle subtended at the atom centre tends towards linearity for proton and tetrahedrality for the oxygen transfer. A second type (b) corresponds to what has been termed an inverted or non-classical potential, in which the equidistant point for the atom (3') is crucially an energy minimum and not a maximum. Both types of potential have been reported for e.g. the proton transfer between the two oxygen atoms of malondialdehyde,1 (4) which implies that the form of the potential is sensitive to factors such as whether electron correlation in the wavefunctions used to evaluate the potentials, and the distance and angle between the proton donor and acceptor atoms.
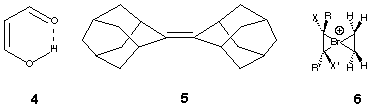
There have been few other reports of atom transfer reactions involving other elements. Brown and co-workers3 have focused on bromination reagents. Calculations by them based on ethene as alkene led to the suggestion that the transfer of a Br+ atom from the halonium ion complex deriving from adamantylideneadamantane 5 to another alkene might proceed through a spiro transition state corresponding to 3, involving a double minimum potential of type (a).4 The possibility of the type (b) mechanism was not evaluated in their studies.
Our own theoretical studies5 on extrusion reactions involving neutral R3I and R3Br species indicated that e.g. alkylated R4I+ or R4Br+ might be viable. We realised an analogy between such species and the type (b) models shown above. Here we investigate this analogy by reporting the results of a study at the ab initio Hartree-Fock (RHF) and correlated (MP2, B3LYP) computational level for the prototypic bromonium ion transfer reaction between two ethene molecules and for substituted and strained derivatives (Scheme 1)
At the Hartree-Fock all-electron level, we obtain6 (Table 1, first four rows) very similar results to those previously reported by Brown.3 The weak complex 2 (Scheme 1) has the bromine atom asymmetrically disposed between the two alkenes analogous to the classic hydrogen bonded complex formed prior to proton transfer reactions. As the quality of the basis set increases, the interaction energy relative to 1 decreases due in part at least to increasing elimination of basis-set-superposition errors. The barrier for the bromine transfer via transition state 3 relative to 2 is about 10 kcal mol-1 at a triple zeta basis level7, with the single imaginary frequency corresponding to 225 cm-1. At this level, the transition state of tetrahedral symmetry 3 is higher in energy than the uncomplexed reactants 1, indicating an overall barrier to the transfer reaction.
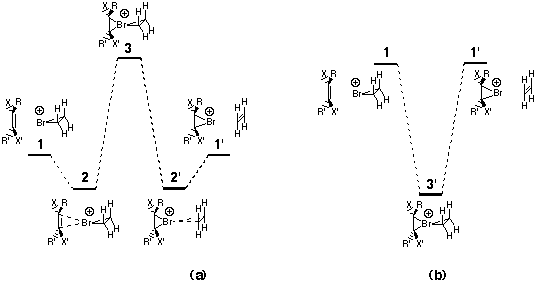
Scheme 1.
Table 1. Energies (Hartree) and energies relative to 1 (kcal mol-1) and transition normal modes (cm-1) for the stationary point structures.
|
Basis Set
|
1
|
2
|
2, rBr-C/Å
|
3
|
3-2
|
3 νi
|
|---|---|---|---|---|---|---|
|
RHF/3-21G(d) |
0.0 (-2715.1822)
|
2.095, 3.159
|
5.3
|
178
|
||
|
RHF/6-31G(d) |
0.0 (-2725.7068)
|
2.060, 3.313
|
10.5
|
236
|
||
|
RHF/6-31(3d,p) |
0.0 (-2725.8840)
|
2.059, 3.429
|
7.5
|
231
|
||
|
RHF/Sadlej pVTZ |
0.0 (-2728.1856)
|
-4.6a
|
2.062, 3.434
|
10.0b
|
225
|
|
|
B3LYP/3-21G(d) |
0.0 (-2718.0589)
|
2.467
|
1.0 |
31
|
||
|
B3LYP/6-31G(3d,p) |
0.0 (-2728.5766)
|
2.459, 2.462
|
0.004
|
95
|
||
|
MP2/6-31G(d) |
0.0 (-2726.3231)
|
2.078, 2.973
|
-8.0
-7.3 (6) |
2.3
|
||
|
B3LYP /Sadlej pVTZ |
0.0 (-2731.0417)
[-2731.1262]e |
-10.7c
[-3.7]e |
2.459, 2.462
|
0.007d
|
75 92, 10 |
aZero point corrected activation energy -4.0 kcal mol-1. Entropy corrected ΔG = +1.9 kcal mol-1. bZero point corrected activation energy 10.3 kcal mol-1; Entropy corrected ΔG = +13.4 kcal mol-1. cZero point corrected activation energy -10.0 kcal mol-1; Entropy corrected ΔG = -0.3 kcal mol-1. dZero point corrected activation energy 0.0 kcal mol-1; Entropy corrected ΔG = -0.9 kcal mol-1.eCorrected for aqueous solvation using the COSMO model (SCRF=(CPCM)).
Noting that the effect of including electron correlation on this potential surface has not been previously reported, we investigated the reaction, initially at the MP2 level (Table 1). Species 2 is now more stable (~10 kcal mol-1) relative to 1. The barrier to bromine transfer from this complex becomes smaller (2.3 kcal mol-1) and the transition state 3 is significantly lower in energy compared to the uncomplexed reactants 1. The computationally faster B3LYP correlated method at the 6-31G(d) and triple-zeta basis set levels reveals an insignificant difference between the energy and geometry of the complex 2 and that of the formal transition state 3. Further correction for zero point energy differences and inclusion of an entropy correction produces a free energy difference of -0.3 kcal/mol between 3/3' and reactants 1. In effect, this now corresponds to a type (b) mechanism in Scheme 1. At this level of theory, we also investigated the geometry involving a 4-coordinate planar central atom 6 (symmetry D2h). Normal mode analysis at this geometry revealed two very small imaginary modes, the larger (92i cm-1) corresponding to bromine transfer, and the smaller (10i cm-1) to rotation of the two ethene units leading towards a tetrahedral geometry. This implies a very flat potential surface in which the transition state 3/intermediate 3' are essentially equivalent, and that interconversion between them requires little energy.
In a series of articles Herges et. al.8 have reported an intriguing class of concerted reactions involving the simultaneous formation of two bonds and breaking of two bonds at a single atom centre, which they classify as co-arctic mechanisms [Scheme 2, (a)]. Such reactions have considerable analogy to pericyclic systems, and are similarly subject to constraints directly equivalent to the Woodward-Hoffmann rules, and which depend on the number of spiro-cyclically conjugated reacting electrons. In the co-arctate classification, the involvement of 4n spiro-cyclic electrons requires a Mobius transition state and requires the co-arctate atom to have a tetrahedral geometry, with an orbital phase inversion occurring at the co-arctate atom. In contrast, 4n+2 electron systems would require a planar geometry at this atom. We note here the evidently strong parallels between such co-arctic systems and the transition states for atom transfer reactions. This parallel also implies that 3' would represent a novel type of co-arctate Mobius system9 existing as an energy minimum rather than as a transition state. Such an analogy also raises the question of whether Br+ transfers should be classified as 4n or 4n+2 coarctate reactions (Scheme 2). The almost identical energies of the tetrahedral and planar coordination around the Br atom suggests that either classification would be equally valid. Indeed, simple electron counting shows that either 6 [4n+2, scheme 2 (b)] or 8 [4n, scheme 2 (c)] electrons could be involved , depending on the role of the lone pair electrons on the bromine, and also to some extent the role of the d-orbitals. We also note that oxygen atom transfers, which clearly proceed through a tetrahedral transition state,2 must according to this scheme correspond to a 4n coarctic reaction, with implied participation of precisely one lone pair on the oxygen atom [e.g. scheme 3 (c) ].
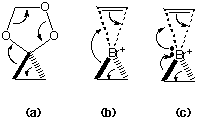
Scheme 2.
The calculations on the parent system reveal that including electron correlation stabilises the four-coordinate mode with respect to the cyclically bridged two-coordinate classical bromonium ion. The latter itself is of course also at one structural limit [Scheme 3, mode (c)] the other corresponding to an acyclic single coordinate Br adjacent to a carbonium ion centre [Scheme 3, mode (a)].
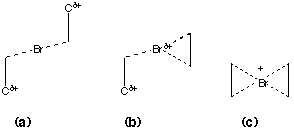
Scheme 3.
Table 2. Energies (Hartree) and energies relative to 1 (kcal mol-1) and transition normal modes (cm-1) for the Substituted Ethene derivaties.
| System | 1 | 2 | 3 | 2' | 1' |
|---|---|---|---|---|---|
| RHF/3-21G* | |||||
| X=CN,X'=H | 0.0 (-2806.4018) | -8.6 | 14.3 (152i) | 14.0 | 21.8 |
| X=OMe,X'=H | 0.0 (-2828.4365) | -8.4 | -7.1 (159i) | -28.2 | -24.3 |
| X=X'=H, R,R'=(CH2)4 | 0.0 (-2869.2092) | - | - | -75.2 | -71.9 |
| X=X'=H, R,R'=(CH2)5 | 0.0 (-2908.0686) | - | - | -47.3 | -41.5 |
| X=X'=H, R,R'=(CH2)6 | 0.0 (-2946.9062) | - | - | -38.0 | -33.6 |
| B3LYP/3-21G* | |||||
| X=CN,X'=H | 0.0 (-2809.7917) | -7.1 | - | - | 16.9 |
| X=OMe,X'=H | 0.0 (-2831.9556) | - | - | -34.1 | -22.1 |
| X=X'=H, R,R'=(CH2)4 | 0.0 (-2873.1901) | - | - | -64.7a | -58.3 |
| X=X'=H, R,R'=(CH2)5 | 0.0 (-2912.3234) | - | - | -48.5 | -40.2 |
| X=X'=H, R,R'=(CH2)6 | 0.0 (-2951.4393) | - | - | -42.1b | -33.1 |
| X=X'=CF3, R,R'=(CH2)6a | 0.0 (-3637.2963) | -15.8 | -15.2 (93i) | -16.1 | 4.5 |
a B3LYP/6-31G* -2884.6524 Hartree. bis (trans cyclohexene) bromonium ion: 3040.6479 b B3LYP/6-31G* -2963.2908, bis (trans cyclooctene) bromonium ion: 3197.9643. c B3LYP/6-31G*. bis (di CF3-trans cyclooctene) bromonium ion: 4546.0517 Hartree.
At the Hartree-Fock level, the substituent X=CN destabilises Br bridging, and hence the bromonium ion derived from unsubstituted alkene 1 is significantly more stable than 1' and the resulting asymmetric complex 2 is in a less deep potential well than the unsubstituted system (Table 2). The cyano substituent also reduces the barrier height 3-2' from 5.3 to 0.3 kcal mol-1. At the B3LYP level, neither 3 nor 2' can be located and the only minimum that appears to exist is 2.
All these trends are reversed when the carbocation stabilising substituent OMe is added. Now, only the complex 2' can be located. It is asymmetric in two respects, firstly in favouring Br binding to the methoxy substituted alkene, and secondly in distorting the bromonium ion towards the single coordinate/carbonium ion extreme noted above [structure (b), scheme 3]. These results suggest that the bromonium cation atom transfer reaction is highly sensitive to the perturbing effects of substituents on the alkenes.
We next investigated how strain on the alkene component could influence the relative energies. We used as our model the three trans cycloalkenes, cyclohexene, heptene and octene (Table 2). We note that such trans cycloalkenes are also chiral, and hence could potentially act as catalysts for the chiral transfer of bromonium cations. The results show clearly that at all levels of theory, strain on the alkene stabilises any bromonium ion deriving from it. This can be easily rationalised by arguing that Br+ coordination changes the carbon hybridisation from sp2 to sp3, thus relieving the strain at those two carbon atoms. The extreme strain in trans cyclohexene also has geometric effects, inducing mode (b), Scheme 3 at the cycloalkene. This is shown to a lesser degree for cycloheptene and not at all for cyclo-octene, which reveals the 4-coordinate mode (c), Scheme 3. As before, the bromine asymmetry also meant that we could not locate the transition states 3, or the adjacent minimum 2.
One can combine the electronic and stain influences to fine tune the properties of such complexes. Thus the electronic effect of the strongly cation destabilising substituent (X=CF3) almost exactly counterbalances the effect of the strained trans cyclo-octene. Here the energies of 1 and 1' are almost identical, and unlike the other systems, location of the transition state 3 is again possible. Its energy relative to 1 is virtually identical to the prototype ethene systems (Table 1), which suggests that at the (computationally too demanding) B3LYP/Sadlej pVTZ higher level of theory it would exhibit similar properties.
Having established that the thermodynamics of such systems can be finely tuned using a combination of strain and electronic effects, we set out to explore in a preliminary manner the possible design of a chiral Br+ catalyst. We focused on the energetics of the equilibrium;
[S...Br...S]+ + [Cat...Br...Cat]+ => 2 [S...Br...Cat]+ (S=substrate, Cat=Catalyst)
The equilibrium needs to favour the right hand side for potential chiral induction by a chiral catalyst, and to reduce the concentration of the [S...Br...S]+ electrophile which would be readily and achirally quenched by nucleophiles. We argued that because of the steric buttressing, the [Cat...Br...Cat] species would not be so quenched, and in the [S...Br...Cat] species, only the desired alkene would be quenched, thus driving the equilibrium over to completion.
The computed equilibrium energy differences (B3LYP/6-31G*) for S=ethene, Cat= trans cyclohexene, cyclooctene and bis(trifluoromethyl) cyclooctene are respectively -37.0, -12.3 and +5.4 kcal/mol (Table 2). This result raises the possibility that a suitably substituted trans cyclo-octene or similar sterically buttressed derivative might be worthy of synthesis to explore whether it might be capable of asymmetric delivery10 of Br+.