A Crystallographic and PM3-COSMO SCF-MOtudy of the Structure and Properties of Aryl Thiazinone Derivatives
ark Hilton[a], Peter Fisk[a],b>Barbara Odell[a], Henry S. Rzepa, [a] Shellesearch Centre, Sittingbourne, Kent. [b] Department ofhemistry, Imperial College, London, SW7 2AY. The solid state structures of threei>p-substituted aryl thiazinone derivatives reveal that the conformationf 2 differs from 1 and 3 in the orientation of the-aryl bond, due in part to an unusually strong C=O...H-C-S intermolecularnteraction of rO...H 2.20Å present in 2. A search of theambridge crystal structure database reveals relatively few other structuresith similarly strong O...H-C interactions in which the common feature is the-C-S structural unit. The observed reaction rate for aqueous hydrolysis ofhis series of compounds via attack of water at the C(2) position wasound to correlate with the energy of the highest unoccupied orbitalorresponding to [[sigma]] rather than [[pi]] character, but no feature inhe wavefunctions could be found which identified 2 as the most likelyo form the strongest O...H-C-S hydrogen bonding interaction in thiseries. As part of arogramme of investigating the properties of cross-conjugated S, N and Oeterocyclic ring systems such as the 2-substituted thiazinone derivativesb>1-3, we required structural information for correlation with theydrolytic reactivity of these systems. A search of the Cambridgerystallographic database for this ring system indicated no entries with thisype of heterocyclic ring system.[1] We thereforendertook a crystallographic investigation of these structures, together with theoretical examination at the semi-empirical PM3 level Experimental Details:-A solution of-chloro-1,3-benzthiazin-4-one[3] and 4-nitrophenol inichloromethane was treated with Et3N added dropwise, the mixture stirredvernight and 2-(4'-nitrophenoxy) 1,3-benzthiazin-4-one collected byiltration; m.p. 225-227[*]C (91%). Found: C, 47.8; H,.5; N, 11.4%. Calcd for C10H6N2O4S: C, 48.0; H, 2.4; N, 11.2%. Similarrocedures gave 2-(4'-nitrophenylthio) 1,3-benzthiazin-4-one, m.p. 179 Crystal Data: 1: C10H6N2O4S, M = 250.2,riclinic, a = 5.547(2), b = 6.383(3), c =.976(4)Å, [[alpha]] = 67.60(2), [[beta]] = 87.00(2), [[gamma]] =1.10(2)[*], V = 258Å[3], spaceroup P1, Z = 1, Dc = 1.61 g cm[-3], Moadiation, [[lambda]] = 0.71073Å, u (Mo-K[[alpha]]) = 3.2 cm Computational Details:- The MOPAC-6. or MOPAC-93 Results and Discussion. Theolecular structures are shown in Figure 1. Two distinct features arebserved in the measured geometries for this series of thiazinoneerivatives. Firstly, the anti S-C(2)-Y-R dihedral conformationa) is preferred for 1 and 3, but the synrientation (b) is found for 2 (Scheme). Secondly, in each casehe aryl and thiazinone rings are essentially orthogonal to each other, with dihedral angles between the aryl and thiazinone rings of 66 and 88 To investigate the origins of the differingonformational behaviour, we inspected each structure for closentermolecular contacts. For 2, we found a strong (2.20Å)ydrogen bonding interaction between a carbonyl oxygen atom of one monomer,nd a H-C(6) bond of another unit (labelled a Figure 2), witho-planarity between the two adjacent thiazinone ring systems. These featuresre found in both crystallographically independent units, with the length ofhe bond derived from each being identical. There are in addition, interlayer...H-C hydrogen bonds (labelled b in Figure 2) that are distinctlyeaker but also present in both independent units (2.40, 2.46Å). Therientation of this interaction disfavours the aryl-Y bond from adoptingonformation (b), and appears to favour the syn orientationa) shown in the Scheme. A much weaker (2.43Å) O...H-C(6)nteraction is also present in 1 (Figure 3), but here the hydrogenond is stepped and the co-planarity of the adjacent linked molecules is noonger present, as a means of avoiding unfavourable intermolecular stericepulsions in orientation (a) (Scheme). Such O...H-C interactions arencreasingly recognised are forming important intermolecular contacts in aariety of systems[5]. A search of the Cambridgerystallographic data base[1] based on the sub-structure...H-C-S in which the O to H distance is < 2.35Å suggests that thispecific interaction is also well established (Table 2), although only twother examples reveal a shorter interaction than in 2. In the instancef thiamine picrate,[6 ]the short (2.21Å) O...H-C-Sydrogen bond can be directly related to the high kinetic acidity of the C(2)roton, which suggests that H-C(6) in 2 might also show similarehaviour. To investigate whether this structural feature related tohe rate of hydrolysis of these derivatives, which involves nucleophilicttack of water at the C(2) position of 1-3, we initially performedM3 semi-empirical calculations at the isolated molecule SCF-MO level foromparison with the crystal structures. In all cases, optimisation convergedo the same orthogonal orientations as determined in the crystal structures.or 2 however, orientation (a) is preferred to (b) by 3cal/mol, providing some measure of support for the orientation in therystal lattice of 2 being brought about by intermolecularnteractions. Inspection of the calculated isolated molecule charge on theydrogen attached to C(6) and the H-C [[sigma]][*]orbitalnergy indicated that compound 1 has a slightly more positive chargen the hydrogen atom (0.146) and the lower energy H-C [[sigma]] We have established from kinetic measurements that theydrolytic half lives at pH9 are approximately 1 (2 hrs), 2< 1 hr) and 3 (30 hours), in each case the products correspondingo attack by water at the C(2) position and subsequent elimination ofhenoxide or thiophenoxide. The PM3 calculations were repeated at theM3/COSMO level in order to provide a more realistic model for the electronictructure of the substrates in aqueous solution, since it has been noted byatritzky and Karelson[7] that orbital energies may beignificantly influenced by the effect of a surrounding polar medium such asater. The lowest unoccupied orbital in all four systems is found to be a[sigma]][*] orbital delocalised over the C(6)-S-C(2)-Yramework, and extending into the [[sigma]] system of the aryl ring. Thisrbital will therefore play a crucial role in accepting electrons from aucleophile such as water. A direct correlation was found with the energy ofhis orbital and the hydrolytic half-life of 1-3 at pH 9 (Table 3),ith 2 showing the lowest [[sigma]]-acceptor energy, and 3 theighest. The interactions with the [[sigma]] system are clearly only possibleith the aryl groups in the orthogonal orientation as found in the crystalattice, which implies therefore that the role of the aryl substituentropagates largely through this [[sigma]] system, rather than acting in any[pi]]-mesomeric sense. Conclusions: The orientation of the-aryl bond in the series of compounds 1-3 is established by X-rayrystallography as corresponding to an orthogonal orientation with respect tohe thiazinone ring system. This results in [[sigma]] rather than[pi]]-conjugation with the bonds associated with C(2), and a directorrelation of the calculated [[sigma]][*] orbitalnergies with the rate of hydrolysis of this type of system. The factorshich promote O...H-C(6) intermolecular hydrogen bonding for 2 onlyemain obscure. We thank Shell Research and the ORS scheme for atudentship (M. Y) and the SERC for diffractometer and computing equipmentrants. eferences. 1. F. H. Allen, J. E. Daview,. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F.itchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Comp. Sci.,991, 31, 187. Release 5.5 of the Quest software was used (March,993). 2. J. J. P. Stewart, J. Comp. Chem., 1989,b>10, 209, 221 implemented in MOPAC-93 from Fujitsu, Tokyo, Japan, andvailable from Quantum Chemistry Program Exchange, University of Indiana,loomington, Indiana. 3. H. Yamamoto, Chem. Pharm.ull., 1983, 31, 1929. 4. A. Klamt and G.hüürmann, J. Chem. Soc., Perkin Transactions II, 1993,99. 5.R. D. Green "Hydrogen Bonding by C-H Groups", Wileynterscience, New York, 1974. R. Taylor, O. Kennard, J. Am. Chem.oc., 1982, 104, 5063; T. Steiner and F.Saenger J. Am. Chem.oc., 1992, 114, 10146. Hydrogen bonding is also known to acidicerminal H-C alkynes ; G. R. Desiraju, J. Chem. Soc., Chem Commun,990, 454. 6. M.-J. Kim, I.-H. Suh, K.Aoki and H.Yamazakii>Acta Cryst.C, (Cr.Str.Comm.), 1988, 44, 725. See also C. L.acLaurin and M.F.Richardson, ibid, 1983, 39, 854. . A. R. Katritzky, M. Karelson, Tetrahedron Letters 1990,b>31, 2987. Captions for Figures.Figure 1. Moleculartructures for (a) 1, (b) one of the pair of crystallographically independentolecules in 2. and (c) 3. Figure 2. Intermolecular contacts in 2, showinghe close approach between the C=O group of one molecule and the H-C(6) ofnother labelled as a and the weaker carbonyl to aryl ring interlayer O..H-Contact labelled b. Figure 3. Intermolecular contacts in 1, showing theoticeably weaker O...H-C interaction and the stepping of adjacent thiazinoneings, compared with 2.
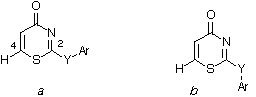{kind=link}