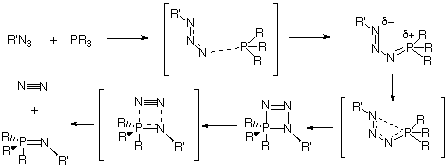
The lowest energy pathway for Staudinger reaction between a phosphane and an azide is predicted at the RHF and DFT ab initio SCF-MO level to proceed via a s-cis intermdiate 7, followed by cyclisation and elimination of N2 to form a phosphazene. When suitable stabilising substituents are present, 7 can instead isomerise to the isolable s-trans intermediate 9. Natural bond orbital perturbation theory analysis has been employed to identify the factors influencing the relative stability of the s-cis phosphazide and the s-trans isomers.

The structure of the phosphazide intermediate 3 was subject of some controversy1d,2 until X-ray structural data became available1d,3. Although they are not usually stable, some phosphazides have been isolated4. Most of these stable phosphazides present distinctive characteristics such as a) the presence of electron withdrawing substituents on the azido end and/or electron donating substituents on the phosphorus atom, b) the presence of very bulky substituents (R and/or R'), and c) the presence of acidic hydrogen atoms capable of forming intramolecular hydrogen bonds with the N atoms of the PN3 unit.
Herein, we present the results of ab initio Hartree-Fock and Density Functional Theory calculations which provide insight into the mechanism of this reaction, and which can offer suitable explanations for some of the experimental facts.
Approximate geometries were initially optimized at the PM3 level using the MOPAC V6.0 program5 implemented on CAChe workstations. The transition structures were located by using eigenvector following routine (TS). This procedure was followed by ab initio calculations performed using the GAUSSIAN94 and 98 program systems6, transition states being located by using the Berny algorithm7 or the synchronous transit-guided quasi-Newton (STQN) methods implemented by Schlegel8. A Natural Bond Orbital (NBO) analysis9 was invoked using the population keyword in GAUSSIAN94/98. Molecular co-ordinates in the form of pdb files for located stationary points are integrated into this article in an enhanced on-line form, together with animations of all important imaginary modes showing the form of the eigenvectors, at the following URL:
http://www.ch.ic.ac.uk/rzepa/RSC/P2/CR3/
Our first aim was to reconcile why almost all the reported X-ray structural data of phosphazides 3 revealed a species with a central N-N bond with the s-trans configuration 9 (Scheme 2), whereas the subsequent extrusion of nitrogen and the formation of an N-P bond requires a s-cis configuration 7.
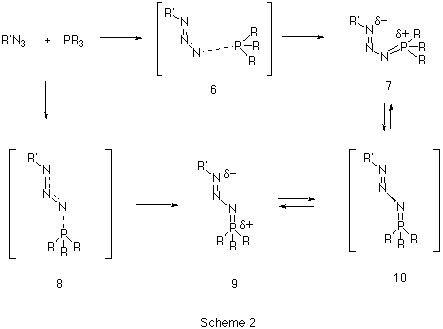
Our results predict the unsubstituted s-cis isomer 7 (R=R'=H) to in fact be more stable10 than the s-trans isomer 9, by 6.6 kcal/mol at the HF level and 9.4 and 8.7 kcal/mol at the B3LYP/6-31G* and 6-311G** levels respectively (Table 1). A simple rationalisation is in terms of favourable electrostatic interactions between the partially positive phosphorus atom (Mulliken charge +0.54/B3LYP/6-31G*) and the partially negatively charged nitrogen atom (-0.37/B3LYP/6-31G*) in the s-cis isomer 7. The geometry of the s-trans isomer also shows a significant double bond character for the N-P bond. This bond distance is longer for the s-cis isomer, which also shows a shortening of the central N-N bond (Figure 1).
A study of the potential energy surface for the formation of 3 revealed that not only was the s-cis isomer 7 route the most stable but also corresponded to the pathway with the lowest energy barrier. The transition state 8 for N-P bond formation has an energy barrier of 61.8 kcal/mol, which is 25.9 kcal/mol larger than the alternative 6 at the RHF level, whilst 8 could not could even be located at the DFT level. A further study of the potential energy surface allowed us to locate another transition structure 10, corresponding to the isomerization of the phosphazide molecule by rotation of the central N-N bond. The location of this transition structure meant that the s-trans isomer could be formed from the s-cis isomer by isomerization,with a readily accessible energy barrier (Table 1) of 20.8 kcal/mol (HF) or 16.46 kcal/mol (B3LYP).
Our results for R=R'=H did not explain why only the s-trans configuration is found for those exceptionally stablised phosphazides which have been isolated. To identify why such stabilization takes a greater effect on the s-trans isomer than on the s-cis, we calculated systems with either a cyano substituent on the nitrogen atom (R'=CN, R=H) or a p electron-donating substituents on the phosphorus atom (R'=H, R=OMe). In the first case, the difference between the energy of the s-cis and s-trans isomers was reduced from 9.4 (R=R'=H) to 5.9 kcal/mol at RHF level. In the second case (R'=H, R=OMe) the s-trans isomer was indeed found to be very slightly more stable than the s-cis isomer at the RHF/6-31G* and B3LYP/6-311G** levels (Table 1). The energy barrier for the formation of the phosphazide for R'=H, R=OMe was still lower for the s-cis isomer. We suggest therefore that the formation of stabilised s-trans isomers takes place via the formation and subsequent isomerisation of the s-cis phosphazide by rotation of the central N-N bond.
In order to get a deeper insight into the factors that control the relative stability of the s-cis and s-trans isomers, we made use of the Natural Bond Orbital (NBO) analysis, a method which has been previously employed in the study of the cis-effect in 1,2-difluoroethene and other organic molecules11. We note firstly that for the case of R=R'=H, the NBO (RHF/6-31G* level) shows a one-centre p lone pair np(Na) on the N atom connected to the P, rather than a two-centre p bond between the Na and P atoms. The most significant difference between the two isomers is that the term corresponding to the interaction np(Na) to p*(Nb=Ng) is 58.8 kcal/mol for the s-trans isomer and 95.5 kcal /mol for the s-cis . The difference is due to the differences in energy between both orbitals ( 0.47 Hartree for s-cis and 0.50 Hartree for s-trans) and in the Fock matrix element ( 0.195 Hartree for s-cis and 0.161 Hartree for s-trans). In addition, the antiperiplanar contributions also favour the s-cis isomer. Thus the antiperiplanar interaction ns( Nb) to s*( Na-P ) is large for the s-cis whereas it is insignificant for synperiplanar s-trans interaction (16.7 kcal/mol vs 0.7 kcal/mol). The interaction ns(Na) to s*(Nb=Ng) is analogous (8.2 kcal/mol vs 0.7 kcal/mol). For the substituted system (R=OMe, R'=H) the NBO shows in contrast a p orbital between Na-P rather than an n lone pair on Na, although the contribution to this orbital is mainly from the N (93%). In this case, the term for the interaction p(Na=P) to p* (Nb=Ng) is 50.2 kcal/mol for the s-trans isomer and 41.75 kcal/mol for the s-cis. The Fock matrix element is the same for both isomers and the difference is due to both orbitals being closer in energy for the s-trans isomer (0.67 Hartree vs. 0.57 Hartree). Although the antiperiplanar contributions are still in favour of the s-cis isomer, e.g. ns( Nb) to s*( Na-P ) is 13.70 kcal/mol for s-cis and 0.5 kcal/mol for s-trans and similarly ns(Na) to s*(Nb=Ng) is 9.2 kcal/mol for s-cis vs 1.0 kcal/mol for s-trans, they do not seem to be enough to make the s-cis isomer more stable.
The second step of the Staudinger reaction takes place when the
s-cis-phosphazide undergoes cyclization to form the
four-membered cyclic intermediate 13 (Scheme 3). Such
cyclization would be expected11 to take place via a
nucleophilic apical attack of the terminal nitrogen atom at one of
the faces of the tetrahedral phosphorus. This mechanism would produce
cycle 12 with the sp3 nitrogen atom in an apical
position. Given the known12 apicophobic character of the
NH2 moiety, this species would evolve via
pseudorotation12,13, to the a priori more stable
isomer 13, in which the sp2 nitrogen atom is in an
apical position because of the increase in apicophilicity of
substituents in pentavalent phosphorus on increasing the
electronegative character. However, the cyclization transition state
11 (Figure 2), was found to link Z-phosphazide 7 with
the cyclic isomer 13 without the intermediacy of 12.
The vibrational analysis of transition structure 11 shows a
rotation of the N-P bond simultaneous to the approach of the nitrogen
and phosphorus atoms. The energy of 11, at least with R=R'=H
is in fact lower than for 10, which explains why the
s-cis-phosphazides are not usually isolated. Subsequently, the
extrusion of a nitrogen molecule from the cyclic intermediate can
take place by fragmentation of the P-N bond in the apical position
(transition state 14) without the need for a pseudo-rotation
process.Again, the energy of 14 is lower than than 10,
confirming that a low energy route does exist for nitrogen extrusion
compared to isomerisation (Figure 3).
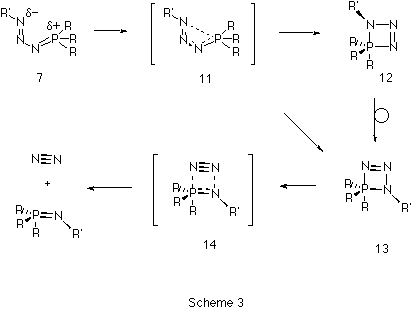
At the ab initio RHF and B3LYP levels of theory, we predict that the Staudinger reaction consists of a sequence of reactions shown diagrammatically in Figure 3. Nucleophilic attack of the phosphane on the terminal nitrogen of an azide forms the corresponding s-cis-phosphazide 7, which in turn can either isomerize to the s-trans isomer 9, which would only be stable and isolable if appropriate substituents are present, or readily cyclize to form the cyclic intermediate 13. Elimination of a nitrogen molecule would form the corresponding phosphazene 4. Results on larger substituted systems of synthetic interest will be reported in a subsequent article.
[1] (a) H. Staudinger, J. Meyer, Helv. Chim. Acta 1919, 2,
635; b) Y. G. Gololobov, L. F. Kasukhin, Tetrahedron
1992, 48, 1353; c) Y. G. Gololobov, I. N. Zhmurova, L.
F. Kasukhin, ibid. 1981, 37, 437; d) A. W.
Johnson, Ylides and Imines of Phosphorus, Wiley, New York, 1993, p.
403.
[2] J.S. Thayer, R.West, Inorg Chem. 1964,
3, 406; J.E. Leffler, U. Honsberg, Y, Tsuno, I. Forsblad,
J. Org. Chem., 1961, 26, 4810.
[3] G.L. Hillhouse, G.V. Goeden, B. L. Haymore, Inorg.
Chem. 1982, 21, 2064.
[4] (a) J. R. Goerlich, M. Farkens, A. Fischer, P. G. Jones,
R. Schmutzler, Z. Anorg. Allg. Chem. 1994, 620, 707; A.
N. Chernega, M. Y. Antipin, Y. T. Struchkov, I. E. Boldeskul, M. P.
Ponomarchuk, L. F. Kasukhin, V. P. Kukhar, Zh. Obshch. Khim.
1984, 54, 1979; A. N. Chernega, M. Y. Antipin, Y. T.
Struchkov, M. P. Ponomarchuk, L. F. Kasukhin, V. P. Kukhar, ibid.
1989, 59, 1256; A. A. Tolmachev, A. N. Kostyuk, E. S. Kozlov, A. P.
Polishchuk, A. N. Chernega, ibid. 1992, 62,
2675; G. L. Hillhouse, G. V. Goeden, B. L. Haymore, Inorg.
Chem. 1982, 21, 2064; C. K. Lowe-Ma, R. A. Nissan,
W. S. Wilson, J. Org. Chem. 1990, 55, 3755; R.
W. Saalfrank, E. Ackermann, M. Fisher, U. Wirth, H. Zimmerman,
Chem. Ber. 1990, 123, 115; J. Kovacs, I Pinter,
M. Kajtar-Peredy, L Somsak, Tetrahedron, 1997,
53, 1541; M. Hirsch-Kuchma, T. Nicholson, A. Davison, W. M.
Davis, A. G. Jones, J. Chem. Soc., Dalton Trans., 1997,
3185; b) P. Molina, C. López-Leonardo, J. Llamas-Botía,
C. Foces-Foces, C. Fernández-Castaño,
Tetrahedron 1996, 52, 9629; c) C. G. Chidester,
J. Szmuszkovicz, D. J. Duchamp, L. G. Laurian, J. P. Freeman, Acta
Crystallogr. Sect. C 1988, 44, 1080;
[5] J.J.P.Stewart, MOPAC, Quantum Chemistry Program Exchange,
University of Indiana, Bloomington, USA, Program 455.
[6] Gaussian program systems available from Gaussian Inc.,
4415 Fifth Avenue, Pittsburgh, PA, USA.
[7] H.B. Schlegel, J.Comp.Chem., 1982,
3, 214.
[8] C. Peng, H.B. Schlegel, Israel J. Chem.,
1993, 33, 449
[9] A.E. Reed, L.A. Curtis, F. Weinhold, Chem. Rev.,
1988, 88, 899
[10] An early and to the best of our knowledge the only
previous quantum mechanics study4b on phosphazides,
reported the Z configuration of
PH3N3CH3 to be 1.6 kcal
mol-1 more stable than the E isomer at HF/6-31G**
level.
[11] P.vR. Scheleyer, M. Kaupp, F. Hampel, M. Bremer, K.
Mislow, J. Am. Chem. Soc. 1992, 114, 6791. T.
Yamamoto and S. Tomodo, Chem. Let., 1997,
1069-1070.
[12] P. Gillespie, F. Ramirez, I. Ugi, D. Marquarding,
Angew. Chem. Int. Ed.Engl. 1973, 12, 91.
[13] D. G. Gilheany on The Chemistry of Organophosphorus
compounds, The chemistry of functional groups series,
editor S. Patai, Vol3, Chapter 1. Wiley & Sons, 1994.
Figure 1. B3LYP/6-311G** geometries for Ground states 9 and 7, and Transition states 6,8 and 10.
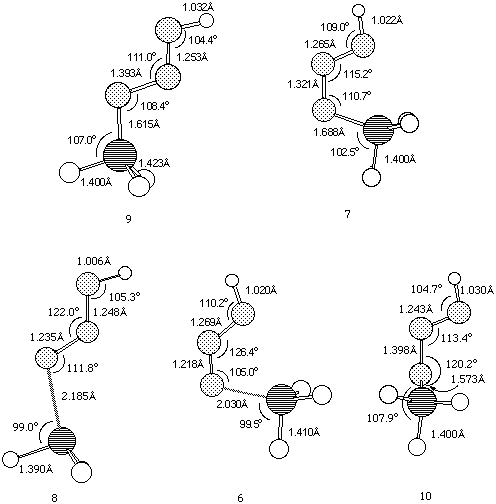
Figure 2. B3LYP/6-311G** geometries for structures
11-14
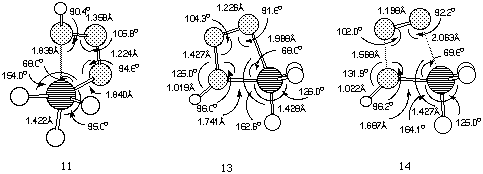
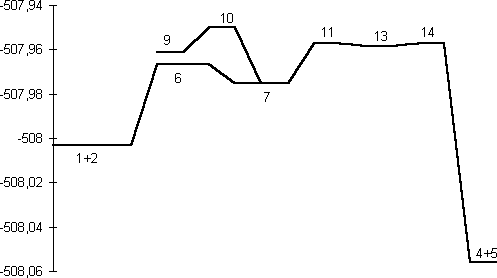
Table 1. Absolute and Relative (to 1+2) Energies and imaginary normal modes for the stationary point structures in the transformation 1+2 to 4+5 (Figure 3).
| RHF/6-31G* | B3LYP/6-31G* | B3LYP/6-311G** | ||||
|---|---|---|---|---|---|---|
|
Energy, Hartree
(Energy relative to 1+2, kcal/mol) |
n, cm-1 |
Energy, Hartree
(Energy relative to 1+2, kcal/mol) |
n, cm-1 |
Energy, Hartree
(Energy relative to 1+2, kcal/mol) |
n, cm-1 | |
|
R=R'=H |
||||||
|
1 |
-342.44795 | -343.14027 | -343.17284 | |||
|
2 |
-163.83869 | -164.78228 | -164.83053 | |||
|
6 |
-506.22902
(36.17) |
-499.4 |
-507.88812
(21.61) |
-325.2 |
-507.96675
(22.98) |
-339.2 |
|
7 |
-506.26216
(15.37) |
-507.89731
(15.83) |
-507.97511
(17.73) |
|||
|
8 |
-506.18812
(61.84) |
-573.8 | ||||
|
9 |
-506.25172
(21.92) |
-507.8818
(25.50) |
-507.96130
(26.40) |
|||
|
10 |
-506.24131
(26.70) |
-176.5 |
-507.87110
(32.29) |
-200.5 |
-507.94970
(33.68) |
-211.4 |
|
11 |
-506.23611
(31.78) |
-350.4 |
-507.88096
(27.1) |
-228.8 |
-507.95697
(29.12) |
-251.2 |
|
13 |
-506.24157
(28.29) |
-507.88159
(25.70) |
|
-507.95829
(28.29) |
||
|
14 |
-506.23536
(32.19) |
-663.2 |
-507.95704
(29.07) |
-542.0 | ||
|
4 |
-397.42731 | -398.44640 | -398.49981 | |||
|
R=OMe, R'=H |
||||||
|
6 |
||||||
|
7 |
||||||
|
8 |
||||||
|
9 |
||||||
|
10 |
||||||
|
R=H, R'=CN |
||||||
|
7 |
||||||
|
9 |
||||||