Method of action
Fluconazole is a fungistatic in action and works by inhibiting the fungal cytochrome P-450 sterol C-14 alpha-demethylation. This is clearly shown below as the fluconazole molecule sits in the active site of the cytochrome therby preventing the lanosterol being oxidised to ergosterol, the principal sterol in the fungal cell membrane. This obstruction of membrane sterol synthesis prevents the fungi from replicating and depletion of ergosterol alters the membrane function and permeability. Another effect which is believed to account for the fungistatic effects of fluconazole is the build up of 14 alpha-methyl sterols. Human, along with other mammalian species contain P450 enzymes which are similar in operation to that of cytochrome P-450 but the important difference making it selectively toxic is in human cells, the major sterol is cholesterol rather than ergosterol, and fluconazole (and other triazoles) have lower incidence of side effects as they are much more specific inhibitors of lanosterol alpha-demethylase.
Cytochrome P-450
Cytochrome P-450 is a name for a family of heme proteins that perform hydroxylation reactions, as well as epoxidation, peroxygenation, desulfuration, dealkylation, deamination, and dehalogenation reactions. The proteins resemble mitochondrial cytochrome oxidase in being able to bind both O2 and carbon monoxide.
Cytochrome P-450 hydroxylates many compounds. These include the hydroxylations of steroid hormone synthesis and the hydroxylation of thousands of xenobiotics (foreign compounds), including drugs such as phenobarbital and environmental carcinogens such as benzpyrene, a constituent of the smoke from tobacco and backyard grills. Hydroxylation of foreign substances usually increases their solubility and is a step in their detoxification, or metabolism and excretion. In some cases, however, some of these reactions activate potentially carcinogenic substances to more reactive species. It is clear that this is vital for the survival fungi so by affecting its operation, fluconazole is an effective antifungal agent.
Pharmokinetics
Fluconazole displays pharmacokinetic properties that are similar following administration by the intravenous or oral routes. In normal volunteers, the bioavailability of oral fluconazole is over 90% compared with intravenous.
The rate and extent of absorption is not affected by the presence of food or stomach acid. Steady-state concentrations are reached within 5 to 10 days following once-daily oral doses. Onset of steady state can occur in as little as 2 days if a loading dose equal to twice the usual daily dose is given.
Protein binding is low (11-12%). The apparent volume of distribution approximates that of total body water. Fluconazole distributes readily throughout the body with good penetration into urine and skin (approximately 10 times the plasma concentration); distribution into saliva, sputum, nails, and vaginal tissue occurs in concentrations approximately equal to that of plasma.
The primary route of elimination is renal, with about 80% of the dose appearing in urine as unchanged drug. Therefore, the pharmacokinetics of fluconazole are markedly affected by a reduction in renal function. About 11% of the dose is excreted in the urine as metabolites. Results of a few limited studies indicate that HIV-infected adults may have a smaller apparent volume of distribution, longer plasma half-life, and reduced renal clearance of fluconazole relative to healthy adults therefore their dose may have to be reduced.
Fluconazole's unique pharmacokinetic profile of having a low molecular weight, low plasma binding affinity, water solubility, little first pass metabolism and long half life is what makes it the drug of choice for treating fungal infections.
In Vitro activity
Fluconazole in vitro activity is extremely poor. Rogers and Galgiani cited in reference ______ have reported that at a physiologic pH fluconazole was 16-fold less active than ketoconazole and amphotericin B against yeasts and filamentous fungi when tested in both liquid and solid medium assays. It was also shown to be active in vitro against Candida species and Cryptococcus neoformans, less active against dermatophytes, and totally inactive against Aspergillus species.
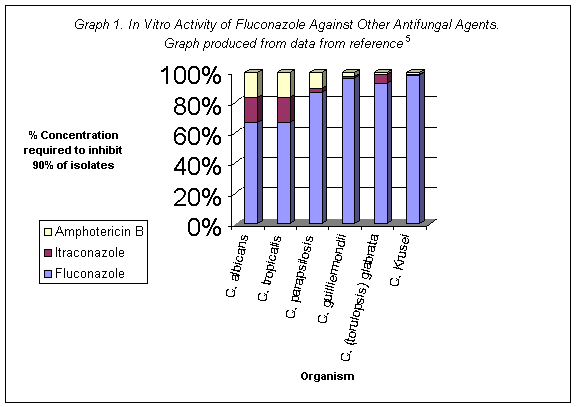
Odds et al. cited in _______examined fluconazole in vitro activity using four different test systems - relative inhibition factors, agar dilution susceptibility assay, effect of drug on hyphal formation of C. albicans and effect on ATP content of C. albicans spheroplasts. Fluconazole was poorly active in all of the assays. Graph 1 shows high doses of fluconazole are needed in comparison to other agents to combat in vitro infection.
In Vivo activity
Fluconazole's poor in vitro activity is in complete contrast to its in vivo activity. Oral administration shows superior activity to ketoconazole and comparable to amphotericin B in treatment of a variety of systemic and superficial fungal infections, including fungal meningitis and Cryptococcus neoformans. Fluconazole was shown to be 20-fold more active in vivo against experimental candidiasis than ketoconazole and is active against tinea versicolor and dermatomycoses. Oral fluconazole has been shown to be active in an animal model of vaginal candidiasis.
Drug Interactions
Due to the specificity of its action, fluconazole has few drug-drug interactions. Several mainstream drugs were tested for adverse effects and these were the findings:
Oral contraceptives there was no significant change in levels of sex hormones after administration of 50 mg daily for 10 days. However higher doses (around 200 mg) have significantly increased levels of circulating levonorgestrel and ethinyl estradiol compared to a placebo
Decreased metabolism of either phenytoin or warfarin
Decreased renal clearance of fluconazole when used with hydrochlorothiazide
Increased hypoglycemia with sulfonylureas
Cimetidine and rifampin decrease flucon level
Rifabutin has increased levels and toxicity
Phenytoin levels may increase, as may zidovudine, delavirdine, and theophylline, oral hypoglycemics of the sulfonylurea type may have increased level, producing hypoglycemia.
May increase sildenafil (Viagra) levels.
DOSAGE AND DIRECTIONS FOR USE
Use in Adults
1. For cryptococcal meningitis the usual dose is 400 mg on the first
day followed by 200 mg once daily. Depending on the clinical response of
the patient this dose may be increased to 400 mg daily. Usually, duration
of treatment for cryptococcal meningitis is 6-8 weeks.
For the prevention of relapse of cryptococcal meningitis in
patients with AIDS, after the patient received a full course of primary
therapy, fluconazole may be administered at a daily dose of at least 100
mg.
2. For systemic candidiasis the usual dose is 400 mg on the first day
followed by 200 mg daily. Depending on the clinical response, the dose
may be increased to 400 mg daily. Duration of treatment is based upon the
clinical response.
Use in Elderly
Where there is no evidence of renal impairment, normal dosage recommendations
should be adopted.
Use in Children
To reconstitute the powder for oral suspension: Tap the bottle to loosen
powder. Add 24 mL of water.
Shake well. Shake immediately prior to use.
As with similar infections in adults, the duration of treatment is
based on the clinical and mycological response. Diflucan is administered
as a single daily dose.
1. The recommended dosage for oropharyngeal candidiasis is 3 mg/kg
daily. A loading dose of 6 mg/kg may be used on the first day to achieve
steady state levels more rapidly.
2. For the treatment of systemic candidiasis and cryptococcal infection,
the recommended dosage is 6-12 mg/kg, depending on the severity of the
disease.
3. For the prevention of fungal infections in immunocompromised patients
considered at risk as a consequence of neutropenia following cytotoxic
chemotherapy or radiotherapy, the dose should be 3-12 mg/kg daily, depending
on the extent and duration of the induced neutropenia.