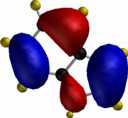
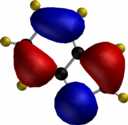
For comparison, the relative energies of the Wheland intermediate resulting from protonation (the model electrophile) are shown, 3-substituent first, 2-substituent second, calculated using the AM1 method.
| furan (3:182.2/2:174.1) | pyrrole (3:199.7/2:200.4) | thiophene (3:199.7/2:200.5) |
|---|---|---|
| |
|
|
| benzofuran (3:192.4/2:189.3) | indole (3:206.7/2:218.2) | benzothiophene (3:205.5/2:214.3) |
| |
|
|
To demonstrate the 3D nature of these orbitals, each thumbnail image below is linked to a 3DMF file. To view these orbital models, you will need a 3DMF viewer such as the Quick3D or 3DMFPlugin (both browser plugins), 3DMF Optimizer (a Macintosh application), Geo3D (for Mac) or 3DMF Viewer for Windows. Windows users must also install the QuickDraw3D libraries from Apple.