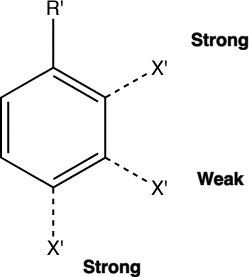
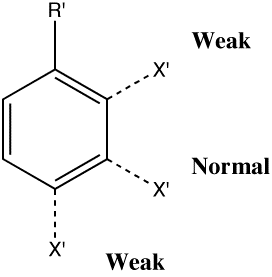
The directing effects of electron donating and withdrawing
groups on electrophilic substitution of benzene
Henry Armstrong, working at Imperial College in 1890, was
the first to categorise substituents (R') on a benzene ring in
terms of the effect they had towards electrophilic substitution
reactions of the ring. With the electron yet to be discovered,
he attributed this to to a polarizable entity he called an
"affinity", and suggested this acted at a distance over the
whole ring, and which differed in character according to the
nature of the substituents (a difference we nowadays categorize
as electron donating or withdrawing).
Erich Huckel, working some 40 years later, was able to
derive from Schroedinger's equation a simple expression which
predicted how "affinities", now named electrons of course, were polarized (the so-called
Huckel molecular orbital theory). These molecular
orbital functions, as we nowadays refer to them, can be used to
illustrate graphically how both the directing effects and the
activating/deactivating effects operate. To maximise the visual
effect, we are going to use two substituents R' that were not
in fact in Armstrong's original list;
CH2+ for an electron withdrawing group
and CH2- for an electron donating group.
Its worth considering just for a moment why we are not using
more conventional neutral groups (i.e. NO2 and
NH2 respectively) for this illustration. Being
neutral, the latter groups can only polarize the molecule by
separating charge to produce a dipolar species. Such ionic
charge separation always takes a fair bit of energy to achieve
compared to neutral covalent bonding.
In contrast, if the R' group is already ionic, then moving the
charge from one part of the molecule to another takes very
little energy, and hence very little if any increase in energy.
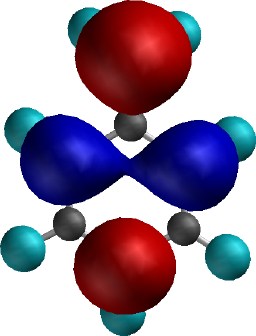 Shapes of (least stable) electron (pair)
deriving from perturbation by "electron donating"
CH2- Substituent. Note how these
electrons have been polarized onto the ortho and
para positions exclusively. An electrophile (electron
loving) reagent will thus be attracted just to those
positions. The energy of this particular electron pair (-1.7eV)
suggests it is only weakly bound by the molecule (most
electrons in molecules have more stable, tightly bound,
energies of -10+ eV), and hence is very reactive (i.e. stabilizing
this electron pair via reaction is likely to be a very
exothermic process). This is conventionally interpreted as
meaning such an electron donating substitutent is highly
activating towards (electrophilic) reaction.
Shapes of (least stable) electron (pair)
deriving from perturbation by "electron donating"
CH2- Substituent. Note how these
electrons have been polarized onto the ortho and
para positions exclusively. An electrophile (electron
loving) reagent will thus be attracted just to those
positions. The energy of this particular electron pair (-1.7eV)
suggests it is only weakly bound by the molecule (most
electrons in molecules have more stable, tightly bound,
energies of -10+ eV), and hence is very reactive (i.e. stabilizing
this electron pair via reaction is likely to be a very
exothermic process). This is conventionally interpreted as
meaning such an electron donating substitutent is highly
activating towards (electrophilic) reaction.
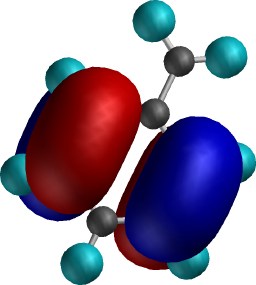
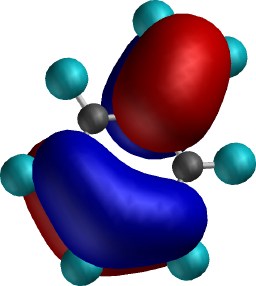 Shapes of (highest and next highest
energy) electrons deriving from perturbation by the "electron
withdrawing" CH2+ Substituent. The
least stable electron pair in this system (left) is nevertheless (due
to the overall positive charge the molecule carries) much more stable with before, with an
energy of about -15.0 eV. This means it is even more stable
than electrons in neutral benzene (~-10 eV), and hence much
less likely to interact with an electrophile (which, in
seeking electrons, does not much like what it sees in such
stabilised electrons) and hence is deactivating. Notice
also that the nodal properties are now quite different from
before. In particular, there is no electron (density) present
on the para position, whereas the meta position
does have it (as does the ortho). Another electron pair (right)
is only slightly different in energy from this first pair
(~-15.6 eV), and this now places electron (density) only on the
meta and para positions, but not the ortho. Taken together then,
these two orbitals both place electrons meta, but
only once each for ortho and para. The latter are
thus not favoured for interaction with an electrophile, and
only the meta position reacts, albeit slowly.
Shapes of (highest and next highest
energy) electrons deriving from perturbation by the "electron
withdrawing" CH2+ Substituent. The
least stable electron pair in this system (left) is nevertheless (due
to the overall positive charge the molecule carries) much more stable with before, with an
energy of about -15.0 eV. This means it is even more stable
than electrons in neutral benzene (~-10 eV), and hence much
less likely to interact with an electrophile (which, in
seeking electrons, does not much like what it sees in such
stabilised electrons) and hence is deactivating. Notice
also that the nodal properties are now quite different from
before. In particular, there is no electron (density) present
on the para position, whereas the meta position
does have it (as does the ortho). Another electron pair (right)
is only slightly different in energy from this first pair
(~-15.6 eV), and this now places electron (density) only on the
meta and para positions, but not the ortho. Taken together then,
these two orbitals both place electrons meta, but
only once each for ortho and para. The latter are
thus not favoured for interaction with an electrophile, and
only the meta position reacts, albeit slowly.