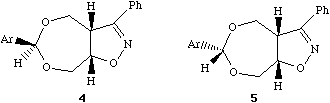
The mechanistic chemists had already established that the critical center was the O-C-O group, in which cleavage of one C-O bond was the important step. In modelling this system, some important early strategic decisions have to be taken. Is the reactivity related to some intermolecular property involving say interaction with water, or is it some structural feature of the molecule itself? Some knowledge of chemistry (the "anomeric" effect) suggests that intramolecular interaction of the C-O bond with the adjacent oxygen electron lone pairs is likely to be important, which may in turn affect say the C-O bond strength and hence its length. This in turn rules out simple molecular mechanics (MM) as the first line of attack, since this is principally a method that does not deal easily with electrons and their wavefunctions. A quantum mechanical (QM) approach should work, but the relative size of the molecule, and its potential conformational complexity implies a major task. Background literature searching and reading shows moreover that QM methods have had a checkered history in dealing with anomeric effects! Its time for a exploratory approach by inspecting the crystal structure.
Using molecular visualisation soon reveals that the solvolytically unstable isomer (4, lhs) has two quite different C-O bond lengths, whereas the relatively stable form (5, rhs) shows almost no difference in these lengths. Thus (4, lhs) is well on the way to breaking a C-O bond, even with no water getting in on the act. Thus this really does appear to be an intramolecular and not an intermolecular problem, where analysing a single structural feature can give great insight into the structure-activity relationships.
Literature Citations. Isoxazolinyldioxepines. Part 1. Structure-Reactivity Studies of the Hydrolysis of Oxazolinyl-dioxepin Derivatives, P. Camilleri, D. Munro, K. Weaver, D. J. Williams, H. S. Rzepa and M. Z. Slawin, J. Chem. Soc. Perkin Trans. 2, 1989, 1929.
But what sort of intermolecular interactions are involved in the binding process? The simple truth is that for the Pirkle reagent, no-one really knew until recently. Intermolecular interactions can be quite difficult to calculate accurately, especially for a molecule that size and the quickest and most direct way to probe this question is to look at the crystal packing structure if it can be measured. A search of the Cambridge crystal structure data base revealed that although no structures between the Pirkle reagent and chiral molecules had been reported, the structure of the racemic reagent itself had already been determined as part of a study on triboluminescence (the crystals emit purple light when you grind them!). The authors conclusion was that a highly unusual hydrogen bond was formed between the -OH group of one molecule and the F3C- group of another. But a small alarm bell sounds at this point. The crystallographers had not actually located the position of the hydrogen atom, they had assumed it could only sit between the O and the F. Could they be mistaken? Visualization enables this assumption to be easily tested.
Literature Citations. A. M. Sweeting and A. L. Rheingold, J. Chem. Phys, 1988, 93, 5648; p-Facial Hydrogen Bonding in the Chiral Resolving agent (S) 2,2,2-Trifluoromethyl-1-(9-Anthryl)ethanol and its Racemic Modification, H.S. Rzepa, M. L. Webb, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 765.
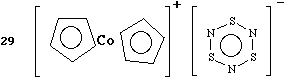
The observation that p-facial hydrogen bonds can form (see above) has a "sensitizing" effect on the perception of this structure! Thus it soon became apparent that this structure also had such interactions, but with some important differences. Here, it is the C-H bonds that interact with the pp orbitals of the SN ring, in itself quite a novel observation. Moreover, each nitrogen supports two such bonds, and significantly, the two hydrogen bonds are almost co-linear. These interactions dominate the chain stacking of the crystal lattice, and hence directly influence both the color and electrical properties of the compound. Best of all, one can see that by adjusting perhaps the size of the metal or the nature of the rings, one might actually improve the co-linearity of the pairs of C-H...N...H-C interactions and hence potentially tune the properties of such systems! Whilst studying crystal structures, be aware that the structure in solution may be quite different! Thus this compound, although black in the solid state, is pale yellow in solution. Moreover, solvation may entirely alter the intra as well as the intermolecular interactions.
Literature Citations. The Preparation, X-Ray structure and Theoretical Study of [CoCp2][S3N3], a Novel Stacking Complex Incorporating Multiple C-H...N(pp) Interactions. P. N. Jagg, P. F. Kelly, H. S. Rzepa, D. J. Williams, J. D. Woollins and W. Wylie, J. Chem. Soc., Chem. Commun., 1991, 942.