Selectively addressable topological templates represent a key feature in the de novo design of proteins using the TASP concept (Template Assembled Synthetic Proteins). We recently described synthetic approaches to regioselectively addressable (orthogonally protected) lysine-containing cyclic decapeptides (RAFT molecules) as templates for TASP synthesis. We now report conformational studies of these molecules by combined NMR and molecular dynamics methods. These results demonstrate the presence of beta-turn and beta-sheet like structures which were predicted for conformationally well-defined templates. Such NMR studies are facilitated by access to molecules with several differentially protected lysines which serve to remove the degeneracy of the sequence.
We also compare the results obtained with purely peptidic templates with those from templates incorporating beta-turn mimetics in order to further refine these structural motifs. The usefulness of this approach to design a priori the structure of such restrained cyclic peptides will be discussed along with potential new applications for RAFT molecules in molecular recognition and combinatorial synthesis.
Introduction
Topological templates which contain spatially defined functional groups are now widely used in protein de novo design and drug discovery1a-j. In principle, any system which can present a fixed array of functionalities may be used, but in the context of de novo design, conformationally constrained peptide motifs are especially interesting. In the TASP approach (Template Assembled Synthetic Proteins)2, amphiphilic peptide secondary structure units are attached to a peptidic template which serves to direct and orient the former into desired folding topologies. To this end, a number of studies from our laboratory have focused on the use of lysine-containing cyclic decapeptides3 of the general form shown in Figure 1. Such molecules were anticipated to provide conformationally well-defined template systems based on two linked type II beta turns, by analogy with systems like the naturally occurring cyclic decapeptide antibiotic, gramicidin S4. These template molecules initially contained only one type of lysine side-chain protection, partly because of the difficulty which exists in preparing cyclopeptides with more than one orthogonal protecting group5 by solution phase peptide methods. However, new applications of these templates, particularly in combinatorial chemistry, require systems with several selectively addressable sites and hence differentially protected side chains. We have recently described a concise route to topological templates of this kind which uses both solid phase and solution peptide synthesis to achieve greater flexibility and efficiency in the preparation of cyclopeptide derivatives with hitherto unprecedented numbers of orthogonal protecting groups or selectively addressable sites. We term such molecules Regioselectively Addressable Functionalized Templates (RAFT)6.
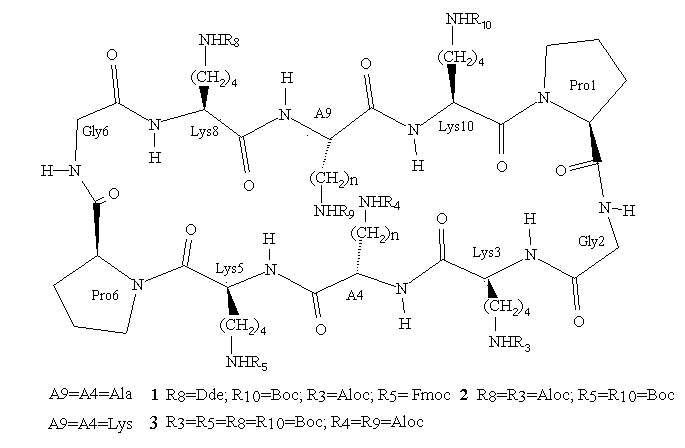
Figure 1.
RAFT molecules (Regioselectively Addressable Functionalized Templates)
represent peptidic templates which contain trifunctional amino acids
whose side chains may be selectively addressed by virtue of orthogonal protection techniques
or a unique chemical reactivity. Ri = addressable function.
As previously noted, the conformations of these molecules were initially inferred from considerations of their amino acid sequence. Apart from one molecular dynamics study7 on closely related peptides there has been no direct study made on the important issue of the solution structure of RAFT. Templates containing only one type of side chain protection are indeed very difficult to study by NMR methods since their high symmetry leads to a considerable loss of information. Now with convenient access to RAFT with distinguishable side chain sites, we have for the first time been able to perform detailed NMR and molecular dynamics conformational investigations, which provide us with valuable information for the continued refinement and application of RAFT in bioorganic chemistry.
Chemistry
The linear decapeptide sequences corresponding to RAFT molecules 1-3 were assembled by solid phase peptide synthesis8 according to the Fmoc strategy9. A highly acid-labile resin linker unit was employed (Sasrin10), thus allowing the completed peptides to be detached from the solid phase resin under very mild conditions, with 1% TFA in DCM. Mild conditions both in the assembly of the peptide and its cleavage from the solid support gave the degree of flexibility needed to incorporate various side chain protecting groups cleavable by acid (Boc), base (Fmoc), Pd(II) (Aloc11), or nucleophiles (Dde12). RAFT 1-3 were subsequently obtained by cyclization of the linear peptides in DMF with PyBOP13 under conditions of high dilution. The cyclization reaction was remarkably efficient, being complete in approximately 30 min with only a slight excess of coupling reagent, and giving the desired RAFT in good to excellent yield after a simple precipitation (see Table 1). These results contrast with those typically observed in peptide cyclizations where long reaction times and excess reagents are normally required14,15, and probably reflect preferred quasi-cyclic conformations16 in the linear sequences for the position of cyclization chosen.
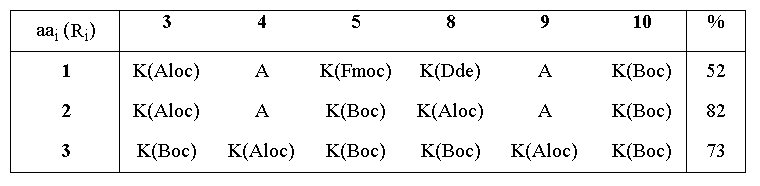
Table 1. Cyclization yields for the synthesis of RAFT molecules
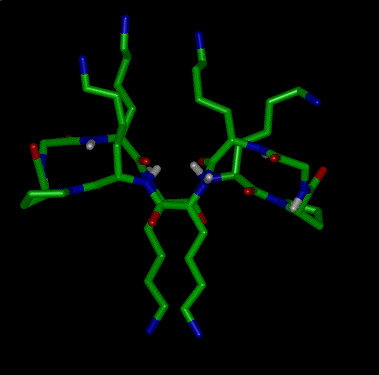
As can be seen from Figure 1, RAFT 1 offers complete differentiation of the four lysine side chains, while 2 and 3 permit binary differentiation, either on the same face (2) or the opposite face (3) of the cycle. Scheme 1 illustrates how these templates are regioselectively addressable in practice for the case of 1. Thus, selective deprotection and acylation as shown permits one to selectively address and modify the four lysine side chains in turn, providing the proper order is respected. The resulting modified RAFT is a potential building block for protein de novo design and TASP synthesis; the newly appended functionalities may be used for the selective attachment of unprotected peptides according to so-called chemoselective ligation methods 17.
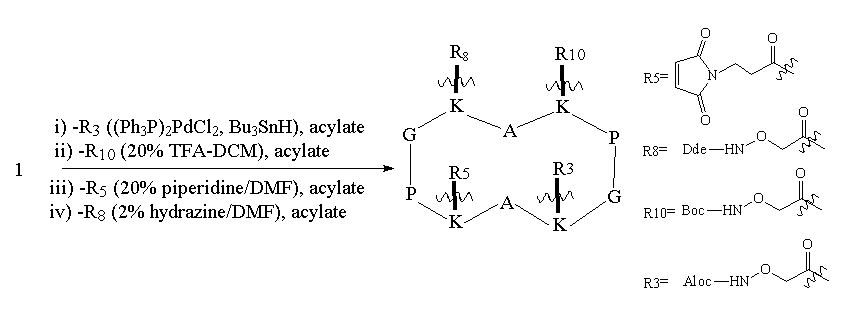
Scheme 1. Regioselective deprotection and modification of RAFT 1
NMR
One and two dimensional NMR spectra were generally recorded in DMSO-d6 on a Bruker AMX600 spectrometer operating at 600.130MHz. Resonance assignments were achieved using DQF-COSY18 and TOCSY19 experiments, and then sequential assignments were extracted by inspection of the dipolar connectivities in the NOESY20-21 spectra. The overall appearance of the 2D spectra for all three compounds was the same as would be anticipated from their structural similarity. Only the spin systems for the two prolyl residues (1,6) were superposed in all three molecules. Indeed, as a result of the differential side chain protection and hence the reduced symmetry in 1-3, two distinct sets of lysine resonances (Lys5 and Lys10, before proline; Lys3 and Lys8, after glycine) could always be distiguished. This is shown very clearly by the fingerprint (NH) region of the NOSEY spectra of 1 and 2 (Figure 2). In the case of compound 1, further removal of symmetry allows yet more information to be extracted, namely the complete identification of all four lysine spin systems rather than just two as in the C2 symmetric templates 2 and 3.
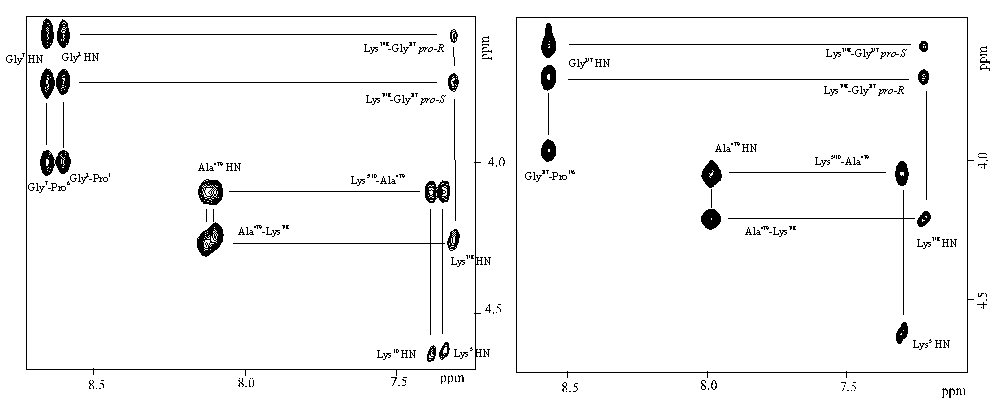
Figure 2. Fingerprint region of the NOESY spectrum of 1 (left) and 2 (right) showing mainly the HN-H(alpha)
dipolar connectivities.
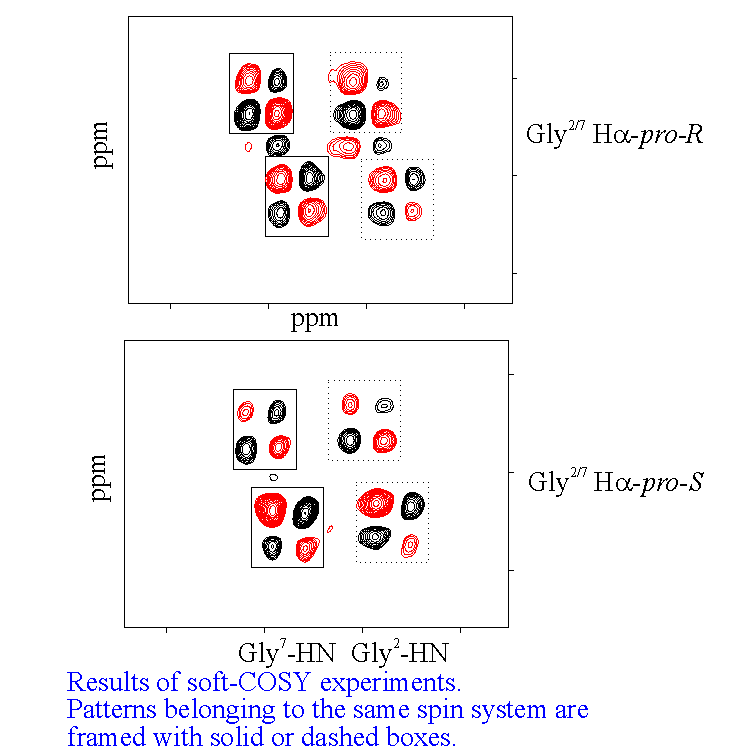
NOE data also demonstrates that all the peptide bonds in the templates are trans. For the prolyl residues, this is shown by an NOE between H(alpha) Lys5/10 and H(delta)Pro6/1. It was also possible to make prochiral assignments for the proline beta and delta protons using the relative interproton NOE intensities between the former and the H(alpha) protons. For the enantiotopic protons of the glycyl residues of 1, a stereospecific assignment was acheived using intra and interresidue NOE data and J coupling values derived from selective (SOFT COSY) experiments22. This permitted us to fix unequivocally the values of the torsion angle psi for the glycyl residues as being around 0 deg. This is the value anticipated for glycine23 in the corner (i+2) position of a type II beta turn.
Further evidence for the location of the Pro-Gly units in a cisoid configuration of a type II turn comes from the appropriate connectivity between the Pro1/6C=O and the Gly 2/7H(alpha) in the HMQC24 spectrum of 1. Also, analysis of the temperature dependence of the amide NH chemical shifts indicate that all residues other than Pro or Gly are participating in hydrogen bonding. This points to an antiparallel beta sheet motif linked at its extremeties by two beta turns, which was the original design principle for our RAFT systems.
CD spectroscopy
A qualitative picture of the secondary structure of 1-3 can also been obtained using CD spectroscopy25. Figure 3 shows the spectra of the three templates recorded in acetonitrile at 22 deg, which indicate that in general terms the former possess the same stable conformation. The form of the CD curves with a negative Cotton effect around 224nm and a positive band around 192-197nm is consistent with the presence in the structures of a type II beta turn. The curves approximate to the form expected for a class B spectrum in the theoretical description of Woody26, although the positive maxima are somewhat blue-shifted compared to the ideal case. Their positions are in fact more typical of those expected for a class A spectrum, which corresponds to a pure beta sheet. Addition of water up to 50% (Figure 3, right) has no significant effect on the spectra recorded in methanol, which perhaps may be regarded as an additional qualitative proof of the conformational stability of these RAFT in solution. Interestingly though, when RAFT 2 is partially deprotected by treatment with acid, the CD spectrum does change appreciably (Figure 4). In methanol-water or water the spectra no longer contain features of both the beta sheet and the beta turn spectra. The positive pi-pi* maximum is red-shifted to around 205-208nm with the negative n-pi* band being essentially unshifted at 224nm, which are the characteristics of the pure beta turn or class B spectrum. This reflects an increase in the motional flexibility of the template and some attendant loss of secondary structure in water (ie, the beta sheet contribution) which is therefore nicely in agreement with the NMR results obtained for this system27.
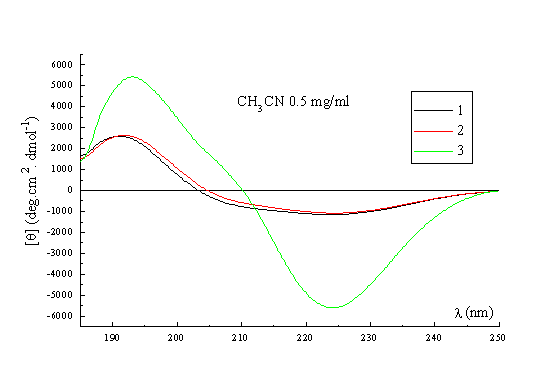
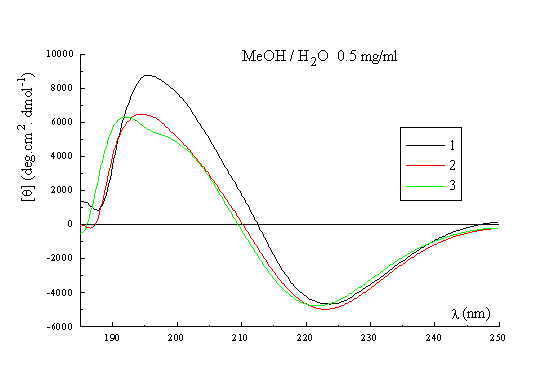
Figure 3. CD spectrum of 1, 2 and 3 in CH3CN (left); CD spectrum of 1, 2 and 3 in MeOH / H2O (right).
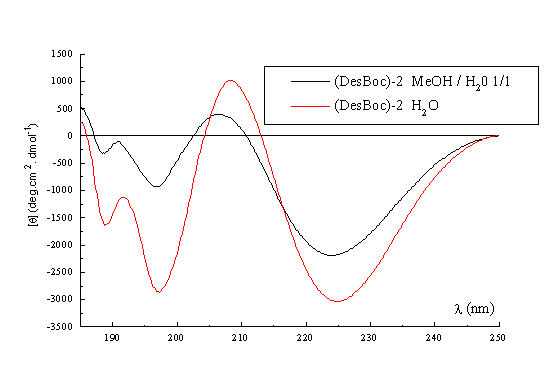
Figure 4. CD spectrum of (DesBoc) -2 in MeOH / H2O, and H2O
Molecular Dynamics
Based on the NMR findings, the starting structure for the molecular dynamics simulations was a system with two Pro-Gly type II beta-turns connected by two Lys-Ala-Lys tripeptide motifs in an extended conformation. The lysine and alanine side chains were directed towards opposite faces of the cycle, which is the configuration originally desired and also that which is consistent with the NMR results. For the simulations with 1, distance constraints evaluated from NOE build-up rates28 were applied chiefly to peptide backbone hydrogens, though lysine alpha-beta separations were also included. In order to avoid errors from coherence transfer effects29, intraresidue NH-H(alpha) distances were calculated from the corresponding 3J NH-H(alpha) coupling constants rather than NOSEY cross peaks30. Finally, as well as the NOE-derived distances, separation bounds were imposed to restrain hydrogen bonds which had been inferred from the NMR analysis.
Simulations were carried out in vacuo using the CVFF force field31 as implemented in Discover (BIOSYM Technologies Inc., San Diego). All the calculations used a distance dependant macroscopic dielectric constant of 2.5*r and an infinite cutoff for nonbonded interactions to partially compensate for the lack of the solvent32. Restrained Molecular Dynamics were performed at 600K or 900K for 100ps using typically a time step of 0.5fs. Configurations were saved and minimized at intervals of 1ps using steepest descent and conjugate gradient algorithms. During the course of a 100ps simulation, no major violations of the experimental distance constraints were observed, the average rmsd for the 43 imposed distances being 0.014 angstroms. Analysis of the backbone dihedral angles (phi, psi) across the dynamics trajectory shows that there is a strong propensity for the extended conformation in the Lys-Ala-Lys units with a slightly distorted type II beta turn about the Pro-Gly locations. The dihedral angle trajectory can be visualized using a linked Ramachandran plot33 (Figure 5). This shows as a function of location number (i), the values of the (phi, psi) pairs linked by straight lines. In this way, a sharp distribution of the (phi, psi) pairs is immediatly evident, which shows that the 100 structures generated in the simulation belong to the same family.
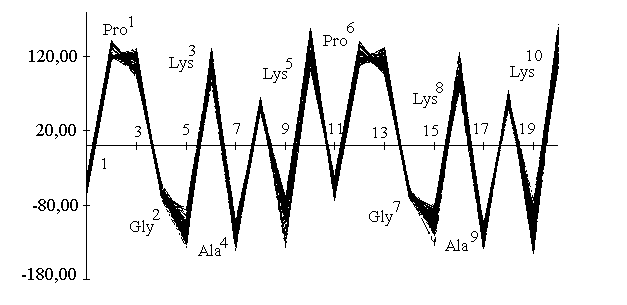
Figure 5. Linked plot representing the variation of dihedral angle (phi, psi,) over the molecular dynamic trajectory as function of the sequence.
The (phi, psi) values obtained for the two prolyl residues (i=1,6) (see Table 2) are characteristic of the i+1 corner position of a type II beta turn (-60, 120). On the other hand, the glycine (phi, psi) values are somewhat distorted with respect to the ideal type II values (80, 0) and correspond rather to those expected for the i+2 position of a type V turn (80, -80).
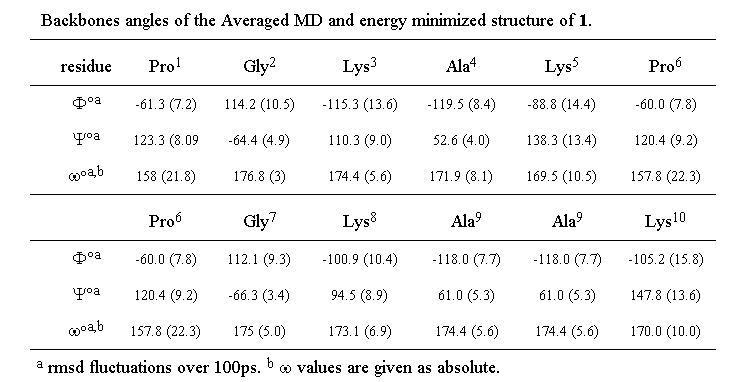
Table 2. Backbones angles of the averaged MD and energy minimized structure of 1
These apparent deviations from ideal type II geometry can be accounted for by variations in the omega values for both prolines which deviate significantly (around 20 deg) from the trans amide value (180 deg) predicted by NMR. Even when a penalty function was imposed in the simulation to keep the proline amide bonds strictly trans, these values persisted. Such a deviation from planarity has previously been reported for the amide bond between hydroxyproline and glycine in a beta turn34. The Lys-Ala-Lys units display angles which correspond to those permitted for a beta sheet structure, though with a certain amount of distortion. This is not altogether surprising since a wide range of structure is permitted within the general beta sheet classification 23 and it would in any case not be realistic to expect two turns and two perfect beta strands to coexist in a cyclic decapeptide without significant strain.
Discussion
Analysis of the experimental data obtained for 1-3 confirm that these RAFT adopt solution structures based on an antiparallel beta sheet conformation, closed by two beta turns centred on the Pro-Gly dipeptides. This is in agreement with the original design principle utilized for the development of such templates. Restrained molecular dynamics using NMR derived constraints support these findings. In particular, the four lysine side chains always remain on the opposite face of the cycle to the two alanines (compounds 1, 2) or lysines (compound 3). These results differ significantly from those of the study of Floegel and Mutter7, in which simulations were performed upon analogous templates containing glycine in the middle of the beta sheet units (ie positions 4 and 9). In this study, several families of favoured conformations were found, some of which had the lysine side chains pointing to both sides of the template in a random fashion.
NMR studies on templates in which the Pro-Gly dipeptides have been replaced by a beta turn mimetic also provide an interesting comparison with our RAFT35. Systems containing the 8-aminomethyl-5,6,7,8-tetrahydro-2-naphtoic acid (AMTA)3 mimetic and glycyl residues in the beta sheet show a much greater flexibility and a tendency for the turn units to fold towards one another. These can be attributed to the replacement of alanine or lysine in our RAFT by glycine which thus leads to a less well-defined conformation that may even permit the inversion of side chains as predicted by Floegel and Mutter7. In view of this, one can conclude that the design of further RAFT molecules should avoid the use of glycine if a conformationally rigid system is to be obtained, and in particular one in which the selectively addressable sites are oriented as desired. Thus, by allowing us to deduce a priori a common secondary structural motif for the backbone of RAFT analogues 1-3, our combined NMR and molecular dynamics approach should permit the design of new RAFT as required while avoiding features which will disrupt the confirmed favoured conformation.
The methods we have elaborated for the synthesis of regioselectively addressable templates may take on added importance now that their conformational preferences and hence the orientation of the side chain functionalities they present have been rigorously determined. Cyclic peptide templates containing four orthogonal protecting groups have already been used as scaffolds for the preparation of combinatorial libraries with a positional scanning format1e,f, for the synthesis and identification of enzyme inhibitors or compounds with antimicrobial activity. Consequently, 1 with four selectively addressable sites may have some useful applications in this context 36. In a similar vein, a wide variety of topological templates, both peptidic and non-peptidic, have been employed to present functional groups in defined orientations for molecular recognition studies, to mimic either receptor binding sites1d or the bioactive conformations of peptide ligands1a,b. RAFT 1 and 2 should be ideally suited for such studies with the scope they offer for the selective attachment of functionalities in a known spatial arrangement36,37. In the area of protein de novo design too, an obvious application for both 1 and 2 would be to attach and orient different peptide blocks giving rise to novel unsymmetrical TASP molecules. Symmetrical TASP derivatives have already been demonstrated to have interesting immunological properties38 and potential for the synthesis of artificial ion channels39, so the availability of RAFT like 1 and 2 should open the way for a wide range of such applications.
Perhaps the most exciting possibilities for the preparation of systems with novel biological or physicochemical properties are offered by RAFT such as 3 in which opposite faces of the molecule are differentiated, thus allowing "effector" and "receptor" functions to be independently presented and modulated on the two faces. This would permit one to include the functions outlined for "monofacial" systems such as 1 and 2 viz a viz molecular recognition and combinatorial techniques, in molecules which can be attached to surfaces or which can interact with cell membranes, via appropriate derivatization of the selectively addressable "lower" face of the RAFT. In the latter case for instance, this might be acheived by grafting lipidic or glycosidic units to the "bifacial" or "two domain" RAFT.
Conclusion
The detailed conformational investigation of RAFT molecules provides valuable information which can be applied in the design of new topological templates and which allows us to refine the role of RAFT in existing applications, as well as offering new perspectives for their usage. These two related themes are currently under active investigation in our laboratory.
Acknowledgement
This work was supported by the Swiss National Science Foundation.
References
1(a) Hirschmann, R.; Nicolau, K. C.; Pietranico, S.; Leahy, E. M.; Salvino, J.; Arison, B.; Cichy, M. A.; Spoors, P. G.; Shakespeare, W. C.; Sprengler, P. A.; Hamley, P.; Smith, A. B.; Reisine, T.; Raynor, K.; Maechler, L.; Donaldson, C.; Vale, W.; Freidinger, R. M.; Cascieri, M. R.; Strader, C. D. J. Am. Chem. Soc. 1993, 115, 12550-12568. (b) Hirschmann, R.; Sprengler, P. A.; Kawasaki, T.; Leahy, J. W.; Shakespeare, W. C.; Smith, A. B. Tetrahedron 1993, 49, 3665-3676. (c) Boyce, R.; Li, G.; Nestler, H. P.; Suenaga, T.; Still, W. C. J. Am. Chem. Soc. 1994, 116, 7955-7956. (d) Kahn, M. Synlett 1993 821-826. (e) Eichler, J.; Lucka, A. W.; Houghten, R. A. Peptide Res. 1994, 7, 300-307. (f) Ostresh, J. M.; Husar, G. M.; Blondelle, S. E.; Doerner, B.; Weber, P. A.; Houghten, R. A. Proc. Natl. Acad. Sci. USA 1994, 91, 11138-11142. (g) Sasaki, T.; Kaiser, E. T. J. Am. Chem. Soc. 1989, 111, 380-381. (h) Liebermann, M.; Sasaki, T. J. Am. Chem. Soc. 1991, 113, 1470-1471. (i) Choma, C. T.; Lear, J. D.; Nelson, M. J.; Dutton, P. L.; Robertson, D. E.; DeGrado, W. F. J. Am. Chem. Soc. 1994, 116, 856-865. (j) Ghadiri, M. R.; Soares, C.; Choi, C. J. Am. Chem. Soc. 1992, 114, 825-831.
2. Mutter, M.; Vuillemier, S. Angew. Chem. Int. Ed. Engl. 1989, 28, 535-554.
3. Ernest, I.; Kalvoda, J.; Sigel, C.; Rihs, G.; Fritz, H.; Blommers, M.; Raschdorf, F.; Francotte, E.; Mutter, M. Helv. Chim. Acta 1993, 76, 1539-1563.
4. DeSantis, P.; Liquori, A. M. Biopolymers 1971, 10, 699-710.
5. Barany, G.; Merrifield, R. B. J. Am. Chem. Soc. 1977, 99, 7363-7365.
6. Dumy, P. Eggleston, I. M.; Cervigni, S.; Sila, U.; Sun, X.; Mutter, M. Tetrahedron Lett. 36, 1255-1258.
7. Floegel, R.; Mutter, M. Biopolymers 1992, 32, 1283-1310.
8. Stewart, J. M.; Young, J. D. 1984 "Solid phase peptide synthesis" (2nd edn.). Pierce Chemical Co., Rockford, Illinois.
9. Atherton, E.; Sheppard, R. C. 1989 "Solid phase peptide synthesis: a practical approach". IRL Press at Oxford University Press.
10. Mergler, M.; Tanner, R.; Gosteli, J.; Gregg, P. Tetrahedron Lett. 1988, 29, 4005-4008.
11. Guibe, F.; Dangles, O.; Balavoine, G.; Loffet, A. Tetrahedron Lett. 1989, 30, 2641-2644.
12. Bycroft, B. W.; Chan, W. C.; Chhabra, S. R.; Hone, N. D. J. Chem. Soc. Chem. Commun. 1993, 778-779.
13. Coste, J.; LeNguyen, D.; Castro, B. Tetrahedron Lett. 1990, 31, 205-208.
14. Kopple, K. J. Pharm. Sci. 1972, 61, 1345-1356.
15. Crusi, E.; Huerta, J. M.; Andreu, D.; Giralt, E. Tetrahedron Lett. 1990, 31, 4191-4194.
16. Mutter, M. J. Am. Chem. Soc. 1977, 99, 8307-8314.
17. Canne, L. E.; Ferre-D'Amare, Burley, S. K.; Kent, S. B. H. J. Am. Chem. Soc. 1995, 117, 2998-3007; Shao, J.; Tam, J. P. J. Am. Chem. Soc. 1995, 117, 3893-3899; Hilvert, D. Chemistry and Biology 1994, 1, 201-203.
18. Piantini, V.; Sorensen, O. W.; Ernst, R. R.; J. Am. Chem. Soc. 1982, 104, 6800-6801.
19. Braunschweiler, L.; Ernst, R. R.; J. Magn. Reson. 1983, 53, 521-528.
20. Jeener, J.; Meier, B. H.; Badimann, P.; Ernst, R. R. J. Chem. Phys. 1979, 71, 4546-4553.
21. Wuthrich, K. 1987 "NMR spectroscopy of proteins and nucleic acids". Wiley, New York.
22. Bruschweiler, R.; Griesinger, C.; Sorensen, O. W.; Ernst, R. R. J. Magn. Reson. 1988, 78, 178-185.
23. Rose, G. D.; Gierasch, L. M.; Smith, J. A. Adv. Protein Chem. 1985, 37, 1-109.
24. Freeman, R.; Morris, G. A. J. Chem. Soc. Chem. Commun. 1978, 684-686.
25. Woody, R. W. 1985 in "The Peptides", Vol.7, pp.16-115. Ed. Hruby, V. J. Academic Press, New York.
26. Woody, R. W. 1974 in "Peptides, polypeptides and proteins", pp.338-350. Eds. Blout, E. R.; Bovey, F. A.; Goodman, M.; Lotan, N. Wiley, New York.
27. When the NMR spectrum of partially deprotected 2 was recorded in D2O, the NOE connectivities characteristic of the beta turn and beta sheet features could no longer be identified. This is consistent with an increase in the structural flexibility of this RAFT molecule.
28. Kumar, A.; Wagner, G.; Ernst, R. R.; Wuthrich, K. J. Am. Chem. Soc. 1981, 103, 3654-3658.
29. Macura, S.; Wuthrich, K.; Ernst, R. R. J. Magn. Reson. 1982, 46, 269-284.
30. Pardi, A.; Billeter, M.; Wuthrich, K. J. Mol. Biol. 1984, 180, 741-751.
31. Hagler, A. T.; Huler, E.; Lifson, S. J. Am. Chem. Soc. 1974, 96, 5319-5327.
32. Weiner, S. J.; Kollman, P. A.; Case, D. A.; Singh, U. C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. J. J. Am. Chem. Soc. 1984, 106, 765-784.
33. MacClain, R. D.; Erickson, B. W. Int. J. Peptide Protein Res. 1995, 45, 272-281.
34. Guba, W.; Haessner, R.; Breipohl, G.; Henke, S.; Knolle, J.; Santagada, V.; Kessler, H. J. Am. Chem. Soc. 1994, 116, 7532-7540.
35. Blommers, M. J. J.; Ernest, I.; Mutter, M. unpublished results.
36. Sila, U.; Mutter, M. J. Mol. Recognition 1995, 8, 29-34.
37. Tuchscherer, G.; Doerner, B.; Sila, U.; Kamber, B.; Mutter, M. Tetrahedron 1993, 49, 3559-3575.
38. Tuchscherer, G.; Servis, C.; Corradin, G.; Blum, U.; Mutter, M. Protein Sci. 1992, 1, 1377-1386.
39. Pawlak, M.; Meseth, U.; Dhanapal, B.; Mutter, M.; Vogel, H. Protein Sci. 1994, 3, 1788-1805.
e-mail to iegglest@ulys.unil.ch
{kind=link}
{kind=link}
{kind=link}