Although serendipity has played an important role in drug discovery [1], computers now are crucial to the rational design of new medicinal agents. The goal of modern computer-aided drug design (CADD) is to make use of statistical and molecular modeling techniques to aid in the design or discovery of new lead structures. It is hoped that computers will help reduce the staggering costs in time and resources that are required to get a new drug to market. The availability of rapid, approximate molecular modeling techniques allow organizations to generate 3D versions of large corporate structural databases with hundreds of thousands of entries [2]. The recent techniques for studying 3D structure-activity relationships (3D-SAR) combine statistical and molecular modeling approaches leading to the generation of very large data arrays [3], which can be managed by chemical structural databases. Furthermore, this technology is being increasingly used by synthetic chemists with little statistical or molecular modeling backgrounds.
The current emphasis in CADD is on lead development, which contrasts with early efforts that concentrated on lead optimization. Techniques for the latter are well-established, while techniques for lead development are still under development and generally involve either (a) using computer technology to propose a new structure to fit a putative or known receptor (de novo design), or (b) searching a database of known structures for those with a desired activity or similarity to active compounds. The latter approach is more widely practiced due to several advantages: (1) Available drugs may already have toxicity and distribution profiles established; (2) the approach makes optimal use of existing resources; and finally, (3) computer software is readily available and does not require modeling expertise to be used effectively. Along with statistical and molecular modeling tools, 3D structural databases have become a key element in modern 3D CADD. This paper describes some 3D searching applications using the ISIS/3D chemical information management system [4].
We should note that the main purpose of 3D searching is to locate potential structures that can fit into a given receptor. Toward this goal, the techniques described in this paper involve developing and optimizing a 3D search query against a database of known compounds. Once an optimum query is obtained (by optimum we mean hitting as many of the active compounds as possible with the fewest possible inactive ones), this query can then be used to search databases of compounds with unknown activities. The resultant hit list will conceivably have a similar percentage of active compounds (or quality leads) as those obtained from searching a database with compounds of known activities.
A key concept in 3D searching is the pharmacophore. The term, introduced by Ehrlich in the early 1900s [5], refers to the molecular framework that carries (phoros) the essential features responsible for a drug's (pharmacon) biological activity. In the current literature, the term has been expanded to refer to the 3D arrangement of functional groups that enable a compound to exert a particular biological effect. Due to stereochemical considerations (i.e., three-point attachments), many pharmacophores are defined simply in terms of three atoms and three distances. If more information is available, other geometric objects and constraints can be added, including constraints on data associated with atoms and bonds [6]. Presently, most pharmacophores are defined in terms of the atoms and bonds of the ligand structures. This ligand-based definition has advantages for input and searching purposes; in the case where the structure of the receptor is completely unknown, it is the only way one can effectively define a pharmacophore model.
It is clear that drugs that bind to a common receptor do not necessarily bind with similar functional groups precisely overlapped; rather, these drugs may show multiple modes of binding [7-9]. The interactions between a drug molecule and a receptor can be dispersed and nonspecific, as in steric and lipophilic interactions - which nevertheless account for most of the free energy changes in binding [10]. Alternatively, they can be highly specific and directional, such as interactions involving H-bond donors and acceptors or those with bound metal ions. But even then, precise overlap of heteroatoms may not result, since the specific interactions occur over a range of angles, distances, and energies [11,12].
Clearly, in such cases, a receptor-based approach to a pharmacophore definition is needed to carry key features of the receptor involved in drug molecule binding. A full receptor-based pharmacophore model complements the ligand-based pharmacophore model for the same activity; it is the 'lock' for a given 'key.' This concept has been expressed and utilized previously, as in the extended molecule approach of Andrews [13]. Our purpose is to show how to implement receptor-based queries in a pharmacophore-based 3D search system. We will also evaluate the success of these queries with respect to their pharmacophoric counterparts by testing them on a database of drugs with known activities.
THE SEARCH PROGRAM: This investigation was carried out using ISIS, chemical information management software which also provides the ability to store and search 3D structures and data. The search capabilities of ISIS/3D include data searching; exact two-dimensional (2D) and 3D matching; 2D and 3D substructure searching, which may include per-atom and atom-pair [6] queries; 3D 'submodel' searching, which allows matching of 3D fragments containing fixed atoms; and 3D conformationally flexible substructure (CFS) searching [14]. Combination of these types of searches allows for the construction of hybrid and flexible queries for specialized applications [15,16].
THE DATABASE: The searches were conducted in MDL Drug Data Report-3D (MDDR-3D) [17] and Comprehensive Medicinal Chemistry-3D (CMC-3D) [18] structural databases. The 94.1 release of MDDR-3D contains 47,926 3D compounds abstracted from the journal Drug Data Report, which contains information from the drug patent literature since 1988 [19]. The 3D structures were built with the CONCORD program of Pearlman [20], and a single conformation is stored in the database.
QUERY DEFINITION: In ISIS/3D, queries are defined using several objects and constraints, which can be selected from a menu. Once added to a structure or query, objects and constraints can be output to standard molecule files. In general, queries will be derived either from the literature or from structures with known activity. In the first case, the 2D substructural features of the query are drawn by the user, who then adds 3D objects and constraints. In the second case, an active structure is brought into ISIS/Draw, the user prunes away the unnecessary portions of the molecule, any atom and bond types are optionally changed, and finally, any other 2D or 3D features are added to create the query. For all searches involving hydrogen or lone-pair atoms, automatic sprouting was conducted at search time, since this information is not stored in the 3D database. Sprouting used standard bond lengths, bond angles, and the perceived valency of the structure atom. The CFS searches are performed with only one difference from the default options: The maximum allowed unique mappings were increased from the default value of 5 to 99.
The models constructed by CONCORD are not necessarily close to the binding conformation in their 3D geometry. This has consequences in 3D searching applications. Many published pharmacophore models are derived from known bound conformations. If a database of CONCORD structures is searched using such a pharmacophore as a query, matches will be found only for the models that happen to be in the correct conformation. The same argument can be made for any database built using computational chemistry techniques.
When searching a database of static conformations, one must modify 3D constraints in published pharmacophores, in terms of the values of the 3D constraints, to hit the correct models in the database. It is therefore important to build the database consistently. Mixing, for example, CONCORD models with models derived from force field calculations or with X-ray crystallographic results could lead to misleading or inefficient pharmacophore queries. As long as a single modeling technique is used in database construction, systematic errors are controlled. The resulting pharmacophore queries may not exactly resemble published ones, but they will be more applicable to the models in a given database.
One approach to dealing with the problem of database consistency is to perform CFS searching, which requires a molecular modeling capability in the searching engine [14,21]. An alternative approach, which can be applied in a static database setting, is to represent conformational flexibility in the search query [16,17]. We now apply searching techniques to a well known area, namely angiotensin converting enzyme (ACE) inhibitors [22].
ACE INHIBITOR DRUG QUERIES: In general, ACE inhibitors contain one or more of the following molecular functionalities:
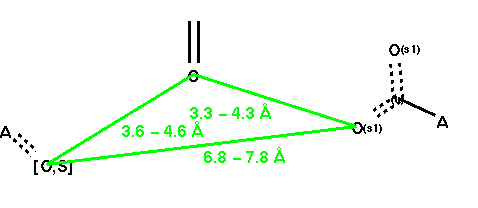
DEVELOPMENT OF 3D QUERIES AND SEARCH RESULTS: The first query, Q-1, displayed in Figure 1, is a simple generalization of the ACE inhibitors query published by Haraki et. al [26]. This was an interpretation of the original Mayer pharmacophore hypothesis [27] with modification on the zinc binding site definition. The carboxylic acid site is generalized as an "any" atom attached to an unsaturated center with two attached, unsubstituted oxygen atoms with a bond definition of S/D (single or double). This guarantees the retrieval of a carboxylic acid where the two oxygens are treated as equivalent due to conjugation. The other side is "any" atom attached with a single or double bond to an oxygen or a sulfur. This will retrieve the known zinc-binding ligand functionalities (i.e., phosphates, sulfides, amides, carbonyls):
The second query, Q-2, is a pseudo-receptor translation of Q-1. In this case, the distances are not given in between the ligand atoms, but are given in between the projected receptor-site points. These points are obtained by projecting a virtual point along the direction of a lone-pair (or hydrogen) at a 3-Å distance from the ligand atom, and surrounded by an exclusion sphere with a 1-Å radius. The distances are tightened to 1-Å tolerance, based on the most rigid ACE inhibitor retrieved by this query. This query may be viewed as a receptor-based query; however it is based on a set of active ligands but not actual receptor-site information (hence the term pseudo-receptor based query).
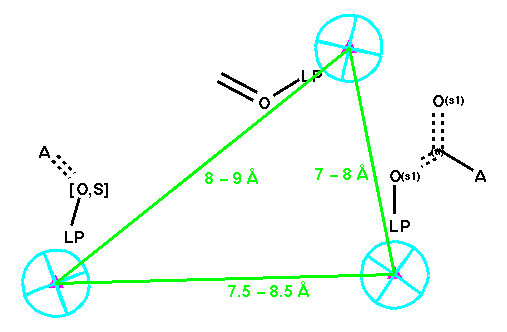
The third query, Q-7, is a mixture of the above: Following Mayer's choice in their use of active analogue approach for ACE inhibitors [27], we used the position of the zinc atom of the ACE as a projected receptor-site point and kept the rest the same as in the ligand-based query.
These three queries were tested against the MDDR-3D database Version 94.1. Of the 47,926 3D structures in this database, 6,761 are listed as antihypertensive and 277 are listed as angiotensinase inhibitors. Most ACE inhibitors fall into one of these categories, but primarily in the latter. Not all of the above listed active compounds contain all of the three functional groups. Hence, a simple substructure search with the fragments that are used in our 3D queries indicated that there are 5,549 eligible compounds in MDDR-3D Version 94.1, of which 677 are listed as antihypertensive and 244 are listed as angiotensinase inhibitors. This constitutes our search domain. Table I summarizes these search results.
| Query | Total no. of hits | Antihypertensives | Angiotensinase inhibitors | |||
|---|---|---|---|---|---|---|
| No. of hits | % Yield | No. of hits | % Yield | % of Actives | ||
| Q-1 | 4,442 | 506 | 11.39 | 231 | 5.20 | 93.15 |
| Q-2 | 5,804 | 623 | 10.74 | 240 | 4.14 | 96.77 |
| Q-3 | 4,473 | 470 | 10.51 | 209 | 4.67 | 84.27 |
The ligand-based query retrieved 4,442 compounds from MDDR-3D, of which 506 were listed under the broad category of "antihypertensives" and 231 were listed under more specific category of "angiotensinase inhibitors." Usually, the latter is a subset of the former. Most of the ACE inhibitors are listed in one (or both) of these two catogories. For practical purposes, we will consider those antihypertensive/angiotensinase inhibitors as the active compounds in the hit list and the rest as false positives. This is only an assumption that was made to have a measurement to compare query performances; otherwise, it is entirely possible that some of the false positives have never been tested for such activity, and they may turn out to be valuable leads.
The "traditional" ligand-based query, Q-1, retrieves a hitlist where 11.39 % are listed as antihypertensive and 5.20 % as angiotensinase inhibitors. Whereas, the pseudo-receptor based query, Q-2, retrieves over a thousand more compounds with 10.74 % as antihypertensives and 4.14 % as angiotensinase inhibitors. While the overall hit list is considerably larger with the receptor-based query, the yield of the listed actives is low. However, considering that there are only 248 eligible angiotensinase inhibitors in the database, the ligand-base query retrieved 231 of the 248 active compounds (93.15 %) while the receptor-based query retrieved 240 (96.77 %) of the eligible active compounds. The mixed query, Q-3, performed slightly worse than the previous two with respect to percent of actives that are retrieved.
Based on the data in Table I, the pseudo-receptor based query, Q-2, retrieved the largest number of compounds; however, with lowest % yield but highest % of eligible actives in the hit list.
Although the crystal structure of ACE is not yet available, its catalytic properties resembles those of two well known zinc proteinases: Carboxypeptidase A and thermolysin. This information has been used extensively to design inhibitors of ACE. Cushman and colleagues designed the first commercial ACE inhibitor from a study of inhibitors of carboxypeptidase A [28]. These researchers reasoned that, since ACE cleaves a terminal dipeptide instead of the single residue cleaved by carboxypeptidase A, the receptor site in ACE must be about 3.4 Å longer, and it should also contain a hydrogen-bond donor and acceptor to bind to the peptide preceeding the substrate's terminal residue. The group designed successively succinyl proline, then its sulfhydryl analog, 3-mercapto-2-methylpropanoyl-L-proline, which became commercially successful (captopril/Squibb). Patchett and co-workers found that the cysteinyl side chain could be replaced by a homophenylalanyl group, yielding inhibitors with reduced side effects (enalapril/Merck) [29].
The terminal phenethyl group in enalapril presumably interacts with a lipophilic region of the ACE receptor, which was a part of the conceptual model of the enzyme developed earlier (site S1). The structure of these inhibitors, when bound to thermolysin, was studied by Monzinga and Matthews, showing a phenylalanine carboxylate bound to the active site zinc ion and the amide nitrogen serving as a hydrogen donor [30]. Hassall and co-workers used molecular modeling to fit captopril to the active site of thermolysin, and then designed a potent bicyclic analog (cilazapril/Roche), which precisely positioned the important functional groups. Their structure-based approach reportedly involved synthesis and screening of fewer than 100 compounds [31].
For this current research, we chose to examine the structures of bound inhibitors to carboxypeptidase A. Structures were obtained from the Protein Data Bank files [32]. The inhibitor/receptor fragments were extracted manually by measuring interatomic distances using ISIS/Draw.
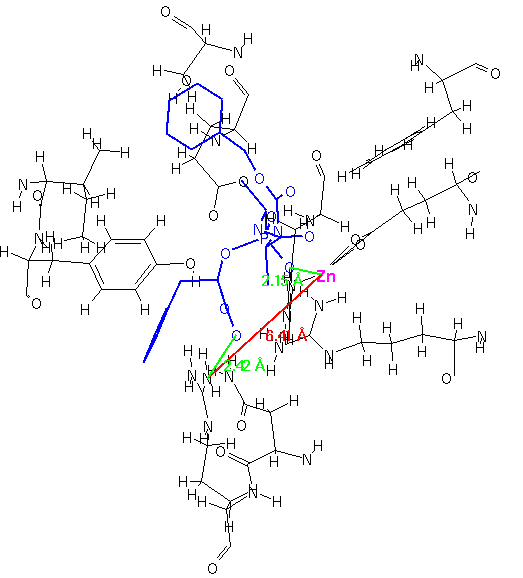
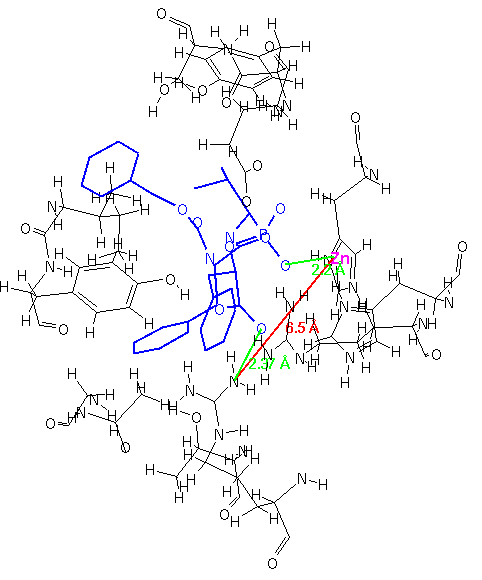
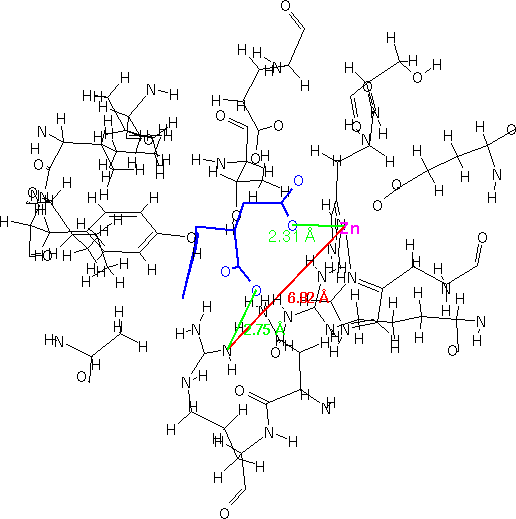
In Figure 4-6, three different inhibitors of the carboxypeptidase are displayed in their bound conformations, as well as the parts of the enzyme that constitute the active site. All three ligands seem to bind in a similar fashion; they also have at least one lipohilic aromatic group. In addition, the two distinct binding sites on the enzyme (shown in red) seem to have consistent distances. Using this information, we can attempt to construct a query: First, we isolate the ligands and the two binding site points from the rest of the enzyme and assign distance ranges that cover all three ligands (see Figure 7).
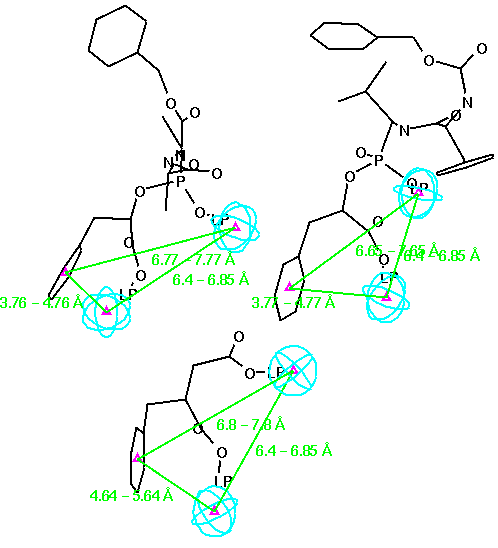
Then we generalize the structural fragments to finalize the initial receptor-based query (see Figure 8).
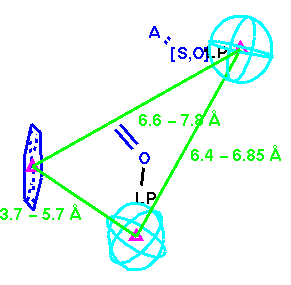
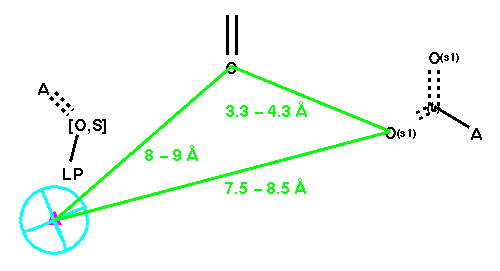
As discussed earlier, existence of the aromatic ring was noted. Hence, we have an opportunity to enhance Mayer's [ligand-based] pharmacophore hypothesis by incorporating the aromatic ring group using the geometric information we gathered from these bound ligands. Furthermore, it was noted earlier that the carboxypeptidase is shorter than ACE. This gives an opportunity to enhance our receptor-based hypothesis by incorporating Mayer's terminal carboxylic acid group (linked with red distance constraints). The composite final query is displayed below (Figure 9).
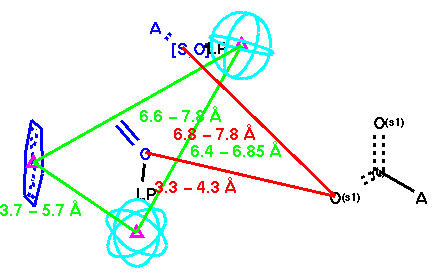
Searching MDDR-3D with Q-4 gives a very large hit list due to the generality of the query. Of the 17,091 compounds in the hit list however, 3,438 are listed as antihypertensives (20.12 %). While the yield for the angiotensinase inhibitors is seemingly low (1.2 %, i.e., 209 of the 17,091 compounds in the hit list) all 209 of the eligible angiotensinase inhibitors (100 %) were retrieved. Hence, the receptor-based query seems to work best but without much selectivity (also consitent with the observation we made with the pesude-receptor based query, Q-2).
Searching the same database (MDDR-3D Version 94.1) with the final query Q-5 gave the results listed in Table II below. Note that out of the 5,041 compounds in MDDR-3D that contain the substructural fragments of Q-5, there are 713 antihypertensive compounds and 193 angiotensinase inhibitors.
| Query | Total no. of hits | Antihypertensives | Angiotensinase inhibitors | |||
|---|---|---|---|---|---|---|
| No. of hits | % Yield | No. of hits | % Yield | % of Actives | ||
| Q-4 | 17,091 | 3,438 | 20.12 | 209 | 1.22 | 100.00 |
| Q-5 | 2,511 | 390 | 15.53 | 176 | 7.01 | 91.19 |
We have emphasized that the fact that a compound is not listed as active does not necessarily mean that it is truly a "false positive" (i.e., a hit that should not have been retrieved). On the contrary, these so-called false positives could be the more interesting compounds due to their potential as new leads. We would like to demonstrate this with an example.
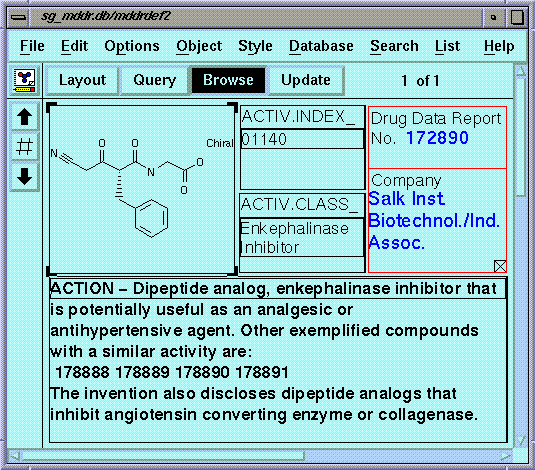
Figure 10 displays an enkephalinase inhibitor that was patented by the Salk Institute in a conformation that was retrieved by performing a CFS search with Q-5.
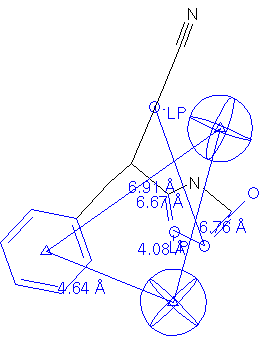
Because of the listed activity, this compound may have been considered a false positive. However in reality, it is expressed in Drug Data Report as "potentially useful" as an analgesic or antihypertensive agent. In fact, it is also reported that several compounds within the discovery are noted to inhibit ACE (see Figure 11). Clearly there may be many other compounds in the database that were never tested for ACE inhibition activity.
As a final test, we ran a search with the final query (Q-5) on the Comprehensive Medicinal Chemistry-3D (CMC-3D) database, which basically contains drugs that are in the marketplace. A search against Version 94.1 of this database retrieved 109 compounds, which included 32 of the 35 eligible known ACE inhibitor drugs. Hence, the query was able to retrieve 91.43 % of the known active compounds, quite consistent with the results obtained from MDDR-3D. We therefore encourage the readers to try and search with these queries in their own databases. The queries are attached as TGF files and can be downloaded to your own desktop with a single mouse click on the figures if ISIS/Draw is set up as a helper application. Please see instructions elsewhere in the ECTOC-1 area on how to set up helper applications.
In this paper, we demonstrated and compared the use of ligand-based and receptor-based queries in 3D substructure searching using ACE inhibitors as an example. We demonstrated the development of pharmacophores making maximal use of simple graphical tools and database information. No modeling skill, hardware, or software was required. This is an important consideration for bench chemist applications.
Presumably there is a balance of forces involved in the binding of an inhibitor to a receptor. Internally, the movement of functional groups in the inhibitor away from global or local energy minima will increase the energy of a given bound conformation. Balancing this are specific and nonspecific interactions with the receptor. A possible strategy, given enough information about the ligand-receptor interaction, would be to develop the ligand-based portion of the pharmacophore in a manner that minimizes the internal energy of the ligand, while developing the receptor-based portion in a manner that optimizes interaction energy and balances the internal energy. We have not yet tried this approach.
Binding and recognition are just half of the equation. Absorption, transport, metabolism, and excretion play huge roles in drug efficacy, and pharmacophore models do not usually take these into consideration. It is possible that combining molecule database searches with searches of metabolite databases [33] could further refine candidate hit lists.
It is possible that searching over a database of structures determined from X-ray crystallography yields hits that are more relevant physically, since crystal packing forces resemble receptor-site interactions somewhat. Ideally, one would search a database of drugs in their bound conformations, but such databases of drug molecules do not exist. In addition, only a relative handful of bound inhibitors have been published, though the numbers are growing. Therefore, we developed a 3D querying strategy that utilizes the available information from receptor-sites with bound ligands, as well as information that can be gathered from the patterns recognized from a list of known active compounds.
For the example we studied, we found that a final composite query (i.e., combined ligand-based and receptor-based query features) improved the search yield, perhaps by accomodating multiple binding modes or variations in specific interactions with the receptor. The yields were only slightly increased over purely ligand-based or receptor-based queries. If the difference is real, however, it would be of considerable significance in reducing the number of candidate structures that need to be evaluated in screening for biological activity.
[1] G. B. Kauffman, Today's Chemist, 13-15 (1989).
[2] A. Rusinko III, R. P. Sheridan, R. Nilakantan, K. S. Nilakantan, N. Bauman, and R. Venkataraghavan, J. Chem. Inf. Comput. Sci. 29, 251-255 (1989).
[3] M. Clark, R. D. Cramer III, D. M. Jones, D. E. Patterson, and P. E. Simeroth, Tetrahedron Comput. Methodol. 3, 47-59 (1990).
[4] ISIS is available from MDL Infromtation Systems, Inc. San Leandro, CA. In this research, the ISIS/Draw and ISIS/Base Versions 1.2, ISIS/Host Version 1.3.1 for SGI IRIX was used.
[5] P. Ehrlich, Dtsch. Chem. Ges. 42, 17 (1909).
[6] O. F. Güner, D. W. Hughes, and L. M. Dumont, J. Chem. Inf. Comput. Sci. 31, 408-414 (1991).
[7] D. H. Rich, C.- Q. Sun, J. V. N. Vara Prasad, A. Pathiasseril, M. V. Toth, G. R. Marshall, M. Clare, R. A. Mueller, and K. Houseman, J. Med. Chem. 34, 1225-1228 (1991).
[8] G. R. Marshall, in Comprehensive Medicinal Chemistry, (Volume 4 - Quantitative Drug Design, C. Hansch, Ed., Pergamon, London, 1990, 446-456).
[9] E. Meyer, J. Mol. Biol 189, 533 (1986).
[10] A. Fersht, Trends in the Biochemical Sci. 9, 145 (1984).
[11] M. Tintelnot and P. Andrews, J. Comput. Aid. Molec. Des. 3, 67-84 (1989).
[12] R. Taylor, O. Kennard, and W. Verschiel, J. Am. Chem. Soc. 105, 5761-5766 (1983).
[13] P. R. Andrews, G. Quint, D. A. Winkler, D. Richardson, M. Sadek, and T. H. Spurling, J. Mol. Graphics 7, 138-145 (1989).
[14] T. E. Moock, D. R. Henry, A. G. Ozkabak, and M. Alamgir, J. Chem. Inf. Comput. Sci. 34, 184-189 (1994).
[15] O. F. Güner, D. R. Henry, and R. S. Pearlman, J. Chem. Inf. Comput. Sci., 32, 101-109 (1992).
[16] O. F. Güner, D. R. Henry, T. E. Moock, and R. S. Pearlman, Tetrahedron Comput. Methodol., 3 (6C), 557-563 (1992).
[17] MDDR-3D, Version 94.1, is available from MDL Information Systems, Inc, San Leandro, CA.
[18] CMC-3D, Version 94.1, is available from MDL Information Systems, Inc, San Leandro, CA.
[19] (a) D. R. Henry, P. J. McHale, B. D. Christie, and D. Hillman, Tetrahedron Comput. Meth., 4(6C), 531-536 (1992). (b) G. Grethe and L. M. Dumont, Drugs News & Persp. 2, 488-490 (1989).
[20] (a) Pearlman, R. S., Chem. Des. Auto. News, 2, 1-7 (1987). (b) CONCORD, Revision 2.9.1, Tripos Associates, St. Louis, MO.
[21] (a) N. W. Murall, and E. K. Davies, J. Chem. Inf. Comput. Sci. 30, 312-316 (1990). (b) T. HUSRT, J. Chem. Inf. Comput. Sci. 34, 190-196 (1994).
[22] D. G. Hangauer, Chapter 7 in Computer-Aided Drug Design, Methods and Applications,T. J. Perun and C. L. Propst, Eds., Marcel Dekker, Inc., New York, pp. 235-295, 1989).
[23] E. W. Petrillo Jr. and M. A. Ondetti, Med. Res. Rev. 2, 1-41 (1982).
[24] P. R. Andrews, J. M. Carson, A. Caselli, M. J. Spark, and R. Woods, J. Med. Chem. 28, 393-399 (1985).
[25] M. R. Saunders, M. S. Tute, and G. A. Webb, J. Comput.-Aided Mol. Des. 1, 133-142 (1987).
[26] K. S. Haraki, R. P. Sheridan, R. Venkataraghavan, D. A. Dunn, and R. McCulloch, Tetrahedron Comput. Methodol., 3(6C), 565-573 (1992).
[27] D. Mayer, C. B. Naylor, I. Motoc, and G. R. Marshall, J. Comput.-Aided Molec. Design, 1, 3-16 (1987).
[28] D. W. Cushman, H. S. Cheung, E. F. Sabo, and M. A. Ondetti, Biochemistry, 16, 5484-91 (1977).
[29] A. A. Patchett, E. Harris, E. W. Tristram, M. J. Wyvratt, M. T. Wu, D. Taub, E. R. Peterson, T. J. Ikeler, J. Ten Broeke, L. G. Payne, D. L. Ondeyka, E. D. Thorsett, W. J. Greenlee, N. S. Lohr, R. D. Hoffsomer, H. Joshua, W. V. Ruyle, J. W. Rothrock, S. D. Aster, A. L. Maycock, F. M. Robinson, and R. Hirschmann, Nature, 288, 280-283 (1980).
[30] A. F. Monzinga and B. W. Matthews, Biochemistry, 23, 5724-5729 (1984).
[31] M. R. Attwood, C. H. Hassall, A. Krohn, G. Lawton, and S. Redshaw, J. Chem. Soc. Pertin Trans. I, 1011-1019, (1986).
[32] Protein Data Bank, January 1995 release, files 1AZM, 1BCD, 1BIC, 1BLC, 1BZM, 1CBX, 1CIL, 1CIN, 1CPS, 1CVA, 2CTC, 6CPA, 7CPA, AND 8CPA. Available from the Chemistry Department, Bldg. 555, Brookhaven National Laboratory, P.O. Box 5000, Upton, NY 11973-5000, USA. Internet: pdb@bnl.gov.
[33] Metabolite, Version 94.2, is available from MDL Information Systems, Inc. San Leandro, CA.